Detection of Helminth Ova in Wastewater Using Recombinase Polymerase Amplification Coupled to Lateral Flow Strips

Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Ascaris Ova
2.2. DNA Extraction
2.3. Design and Screening of RPA Primers
2.4. Optimisation of Lateral Flow RPA Probe
2.5. ITS RPA-LF Amplicon Detection
2.6. Reaction Time and Amplification Temperature
2.7. Detection Limit and Specificity
2.8. Multiplex RPA-LF to Detect Two Different Helminth Ova Genera in Wastewater
3. Results
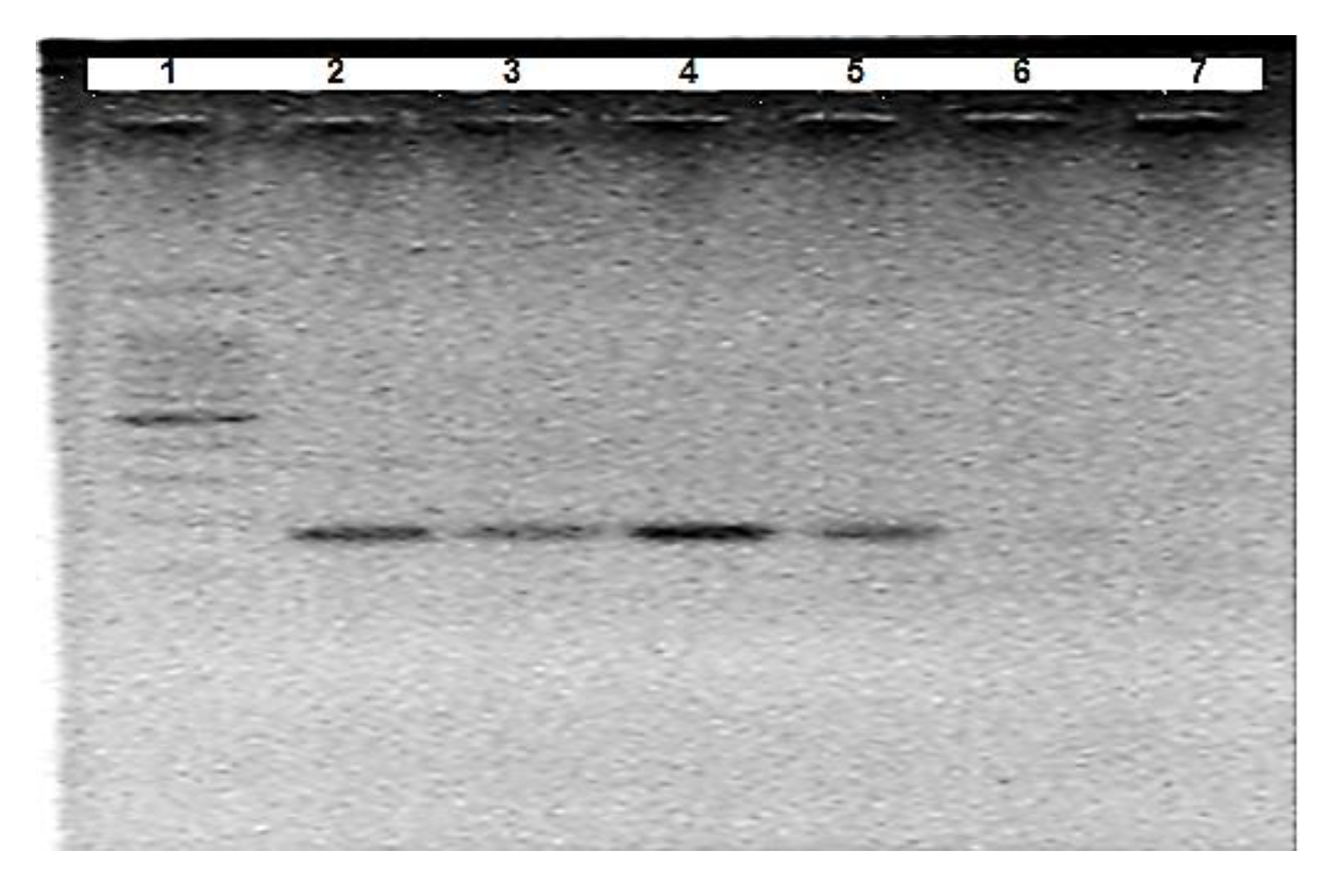
3.1. Optimisation of A. Suum Primers (Asc718F/Asc881R)
3.2. Evaluation of Reaction Time and Amplification Temperature
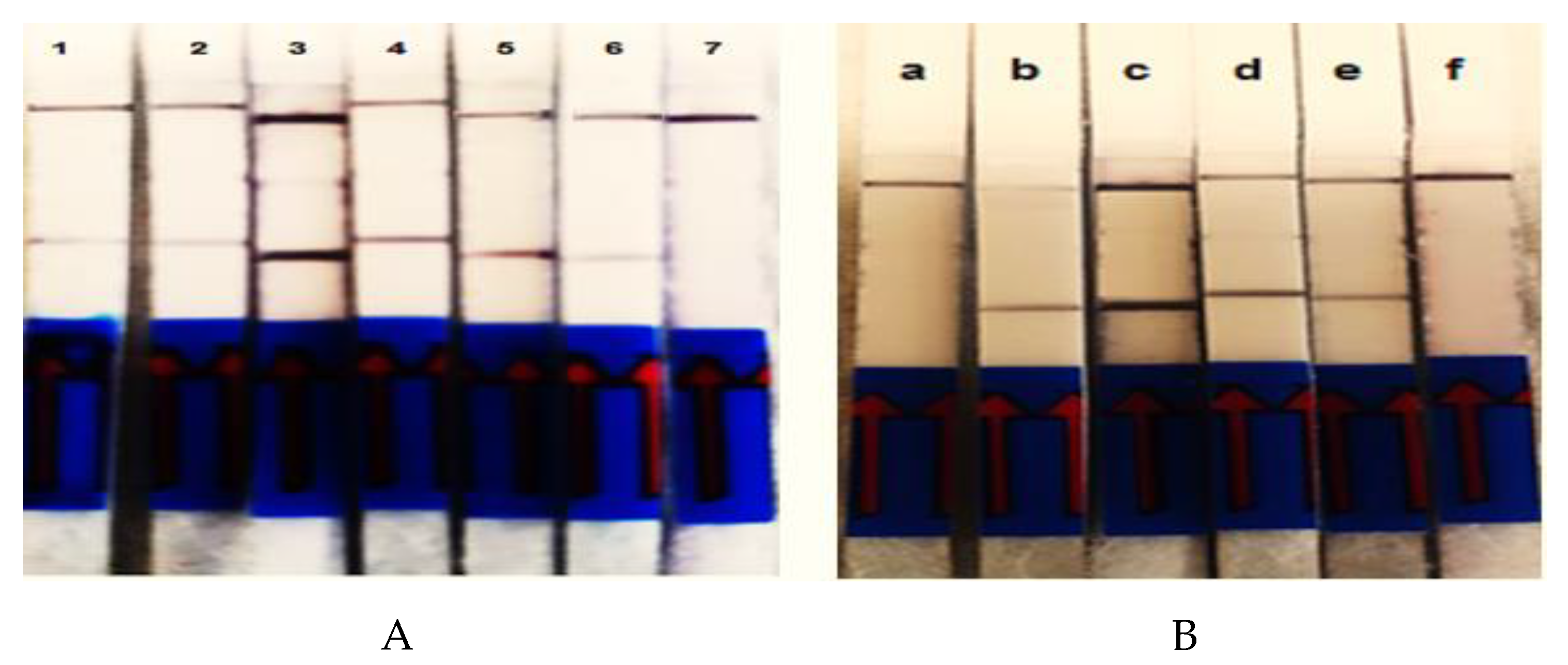
3.3. Evaluation of Detection Limit and Specificity
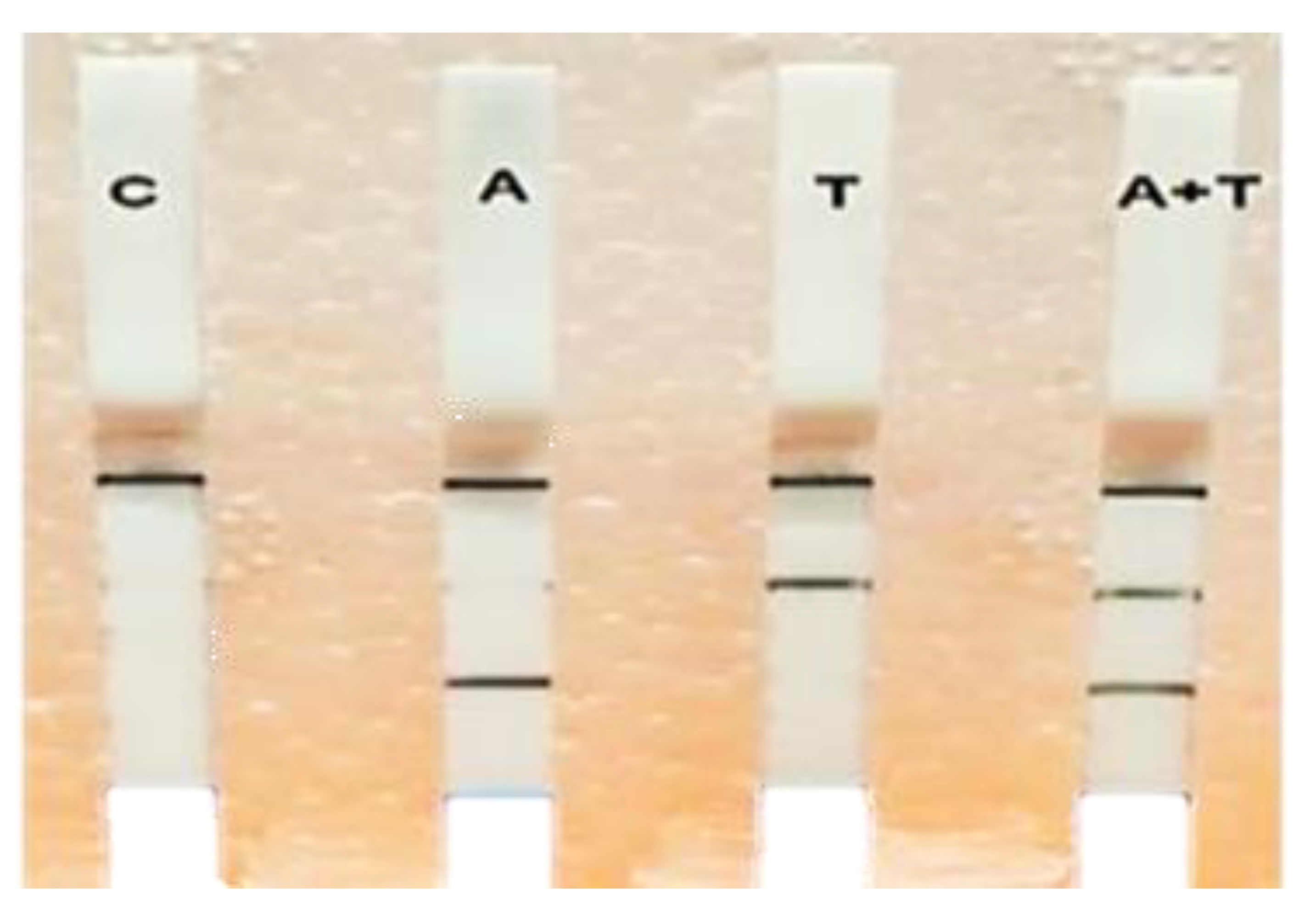
3.4. Multiplex RPA-LF Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rocha, M.C.V.D.; Barés, M.E.; Braga, M.C.B. Quantification of viable helminth eggs in samples of sewage sludge. Water Res. 2016, 103, 245–255. [Google Scholar] [CrossRef]
- Acosta Soto, L.; Santísima-Trinidad, A.B.; Bornay-Llinares, F.J.; Martín González, M.; Pascual Valero, J.A.; Ros Muñoz, M. Quantitative PCR and Digital PCR for Detection of Ascaris lumbricoides Eggs in Reclaimed Water. Biomed. Res. Int. 2017, 2017, 9. [Google Scholar] [CrossRef]
- Gyawali, P.; Sidhu, J.P.; Ahmed, W.; Jagals, P.; Toze, S. Rapid concentration and sensitive detection of hookworm ova from wastewater matrices using a real-time PCR method. Exp. Parasitol. 2015, 159, 5–12. [Google Scholar] [CrossRef]
- Gyawali, P. Infectious helminth ova in wastewater and sludge: A review on public health issues and current quantification practices. Water Sci. Technol. A J. Int. Assoc. Water Pollut. Res. 2018, 77, 1048–1061. [Google Scholar] [CrossRef]
- Li, D.; Tong, T.; Zeng, S.; Lin, Y.; Wu, S.; He, M. Quantification of viable bacteria in wastewater treatment plants by using propidium monoazide combined with quantitative PCR (PMA-qPCR). J. Environ. Sci. 2014, 26, 299–306. [Google Scholar] [CrossRef]
- Collender, P.A.; Kirby, A.E.; Addiss, D.G.; Freeman, M.C.; Remais, J.V. Methods for Quantification of Soil-Transmitted Helminths in Environmental Media: Current Techniques and Recent Advances. Trends Parasitol. 2015, 31, 625–639. [Google Scholar] [CrossRef]
- Amoah, I.D.; Singh, G.; Stenström, T.A.; Reddy, P. Detection and quantification of soil-transmitted helminths in environmental samples: A review of current state-of-the-art and future perspectives. Acta Trop. 2017, 169, 187–201. [Google Scholar] [CrossRef]
- Bethony, J.; Brooker, S.; Albonico, M.; Geiger, S.M.; Loukas, A.; Diemert, D.; Hotez, P.J. Soil-transmitted helminth infections: Ascariasis, trichuriasis, and hookworm. Lancet 2006, 367, 1521–1532. [Google Scholar] [CrossRef]
- Hotez, P.J.; Brooker, S.; Bethony, J.M.; Bottazzi, M.E.; Loukas, A.; Xiao, S. Hookworm Infection. N. Engl. J. Med. 2004, 351, 799–807. [Google Scholar] [CrossRef]
- Hotez, P.J.; Damania, A.; Barua, A.; Stanaway, J. The first “London Declaration”: The Commonwealth and its neglected tropical diseases. PLoS Negl. Trop. Dis. 2017, 11, e0005321. [Google Scholar] [CrossRef]
- Du, R.Y.; Stanaway, J.D.; Hotez, P.J. Could violent conflict derail the London Declaration on NTDs? PLoS Negl. Trop. Dis. 2018, 12, e0006136. [Google Scholar] [CrossRef]
- Karkashan, A.; Khallaf, B.; Morris, J.; Thurbon, N.; Rouch, D.; Smith, S.R.; Deighton, M. Comparison of methodologies for enumerating and detecting the viability of Ascaris eggs in sewage sludge by standard incubation-microscopy, the BacLight Live/Dead viability assay and other vital dyes. Water Res. 2015, 68, 533–544. [Google Scholar] [CrossRef]
- Shahsavari, E.; Schmidt, J.; Aburto-Medina, A.; Khallaf, B.; Balakrishnan, V.; Crosbie, N.D.; Surapaneni, A.; Ball, A.S. A modified assay for the enumeration of ascaris eggs in fresh raw sewage. MethodsX 2017, 4, 186–190. [Google Scholar] [CrossRef]
- Mascarini-Serra, L. Prevention of Soil-transmitted Helminth Infection. J. Glob. Infect. Dis. 2011, 3, 175–182. [Google Scholar] [CrossRef]
- Salam, N.; Azam, S. Prevalence and distribution of soil-transmitted helminth infections in India. BMC Public Health 2017, 17, 201. [Google Scholar] [CrossRef]
- Mara, D.; Sleigh, A. Estimation of Ascaris infection risks in children under 15 from the consumption of wastewater-irrigated carrots. J. Water Health 2010, 8, 35–38. [Google Scholar] [CrossRef]
- Schmidlin, T.; Hürlimann, E.; Silué, K.D.; Yapi, R.B.; Houngbedji, C.; Kouadio, B.A.; Acka-Douabélé, C.A.; Kouassi, D.; Ouattara, M.; Zouzou, F.; et al. Effects of hygiene and defecation behavior on helminths and intestinal protozoa infections in Taabo, Côte d’Ivoire. PLoS ONE 2013, 8, e65722. [Google Scholar] [CrossRef]
- Strunz, E.C.; Addiss, D.G.; Stocks, M.E.; Ogden, S.; Utzinger, J.; Freeman, M.C. Water, Sanitation, Hygiene, and Soil-Transmitted Helminth Infection: A Systematic Review and Meta-Analysis. PLoS Med. 2014, 11, e1001620. [Google Scholar] [CrossRef]
- Manser, N.D.; Wald, I.; Ergas, S.J.; Izurieta, R.; Mihelcic, J.R. Assessing the fate of Ascaris suum ova during mesophilic anaerobic digestion. Environ. Sci. Technol. 2015, 49, 3128–3135. [Google Scholar] [CrossRef]
- Cruz, L.M.; Allanson, M.; Kwa, B.; Azizan, A.; Izurieta, R. Morphological Changes of Ascaris spp. Eggs During Their Development Outside the Host. J. Parasitol. 2011, 98, 63–68. [Google Scholar] [CrossRef]
- Bowman, D.D.; Little, M.D.; Reimers, R.S. Precision and accuracy of an assay for detecting Ascaris eggs in various biosolid matrices. Water Res. 2003, 37, 2063–2072. [Google Scholar] [CrossRef]
- Maya, C.; Ortiz, M.; Jimenez, B. Viability of Ascaris and other helminth genera non larval eggs in different conditions of temperature, lime (pH) and humidity. Water Sci. Technol. A J. Int. Assoc. Water Pollut. Res. 2010, 62, 2616–2624. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horiuchi, S.; Uga, S. Modified flotation method, an effective technique for recovering helminth eggs in soil. Parasitol. Int. 2016, 65, 576–579. [Google Scholar] [CrossRef]
- Sá, M.F.D.; Gonçalves, R.A.; Marder, C.; Baldissera, M.D.; Oliveira, C.B.D.; Noll, J.C.G.; Silva, F.; Monteiro, S.G. Adapted Bailenger method improves the rate of Ascaris suum eggs recovery from liquid pig manure compost. Ciência Rural 2017, 47, 1–6. [Google Scholar]
- O’Connor, N.A.; Surapaneni, A.; Smith, D.; Stevens, D. Occurrence and fate of Ascaris lumbricoides ova in biosolids in Victoria, Australia: A human health risk assessment of biosolids storage periods. Water Sci. Technol. A J. Int. Assoc. Water Pollut. Res. 2017, 76, 1332–1346. [Google Scholar] [CrossRef]
- Stevens, D.P.; Surapaneni, A.; Thodupunuri, R.; O’Connor, N.A.; Smith, D. Helminth log reduction values for recycling water from sewage for the protection of human and stock health. Water Res. 2017, 125, 501–511. [Google Scholar] [CrossRef]
- Llewellyn, S.; Inpankaew, T.; Nery, S.V.; Gray, D.J.; Verweij, J.J.; Clements, A.C.; Gomes, S.J.; Traub, R.; McCarthy, J.S. Application of a Multiplex Quantitative PCR to Assess Prevalence and Intensity Of Intestinal Parasite Infections in a Controlled Clinical Trial. PLoS Negl. Trop. Dis. 2016, 10, e0004380. [Google Scholar] [CrossRef]
- Wardell, R.; Clements, A.C.; Lal, A.; Summers, D.; Llewellyn, S.; Campbell, S.J.; McCarthy, J.; Gray, D.J.; Nery, S.V. An environmental assessment and risk map of Ascaris lumbricoides and Necator americanus distributions in Manufahi District, Timor-Leste. PLoS Negl. Trop. Dis. 2017, 11, e0005565. [Google Scholar] [CrossRef]
- Shiraho, E.A.; Eric, A.L.; Mwangi, I.N.; Maina, G.M.; Kinuthia, J.M.; Mutuku, M.W.; Mugambi, R.M.; Mwandi, J.M.; Mkoji, G.M. Development of a Loop Mediated Isothermal Amplification for Diagnosis of Ascaris lumbricoides in Fecal Samples. J. Parasitol. Res. 2016, 2016, 7376207. [Google Scholar] [CrossRef]
- Bastos, V.K.; Cutolo, S.A.; Doria, M.d.C.O.; Razzolini, M.T.P. Detection and quantification of viable Ascaris sp. and other helminth eggs in sewage sludge. Int. J. Environ. Health Res. 2013, 23, 352–362. [Google Scholar] [CrossRef]
- Pecson, B.M.; Barrios, J.A.; Johnson, D.R.; Nelson, K.L. A Real-Time PCR Method for Quantifying Viable Ascaris Eggs Using the First Internally Transcribed Spacer Region of Ribosomal DNA. Appl. Environ. Microbiol. 2006, 72, 7864–7872. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.A.; McManus, D.P.; Acosta, L.P.; Olveda, R.M.; Williams, G.M.; Ross, A.G.; Gray, D.J.; Gobert, G.N. Multiplex real-time PCR monitoring of intestinal helminths in humans reveals widespread polyparasitism in Northern Samar, the Philippines. Int. J. Parasitol. 2015, 45, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Drain, P.K.; Hyle, E.P.; Noubary, F.; Freedberg, K.A.; Wilson, D.; Bishai, W.R.; Rodriguez, W.; Bassett, I.V. Diagnostic point-of-care tests in resource-limited settings. Lancet Infect. Dis. 2014, 14, 239–249. [Google Scholar] [CrossRef]
- Mugambi, R.M.; Agola, E.L.; Mwangi, I.N.; Kinyua, J.; Shiraho, E.A.; Mkoji, G.M. Development and evaluation of a Loop Mediated Isothermal Amplification (LAMP) technique for the detection of hookworm (Necator americanus) infection in fecal samples. Parasit Vectors 2015, 8, 574. [Google Scholar] [CrossRef]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef]
- Lau, H.Y.; Botella, J.R. Advanced DNA-Based Point-of-Care Diagnostic Methods for Plant Diseases Detection. Front. Plant Sci. 2017, 8, 2016. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2019, 144, 31–67. [Google Scholar] [CrossRef]
- Yang, Y.; Qin, X.; Song, Y.; Zhang, W.; Hu, G.; Dou, Y.; Li, Y.; Zhang, Z. Development of real-time and lateral flow strip reverse transcription recombinase polymerase Amplification assays for rapid detection of peste des petits ruminants virus. Virol. J. 2017, 14, 24. [Google Scholar] [CrossRef]
- Wang, J.; Liu, L.; Wang, J.; Sun, X.; Yuan, W. Recombinase Polymerase Amplification Assay-A Simple, Fast and Cost-Effective Alternative to Real Time PCR for Specific Detection of Feline Herpesvirus-1. PLoS ONE 2017, 12, e0166903. [Google Scholar] [CrossRef]
- James, A.; Macdonald, J. Recombinase polymerase amplification: Emergence as a critical molecular technology for rapid, low-resource diagnostics. Expert Rev. Mol. Diagn. 2015, 15, 1475–1489. [Google Scholar] [CrossRef]
- Euler, M.; Wang, Y.; Heidenreich, D.; Patel, P.; Strohmeier, O.; Hakenberg, S.; Niedrig, M.; Hufert, F.T.; Weidmann, M. Development of a Panel of Recombinase Polymerase Amplification Assays for Detection of Biothreat Agents. J. Clin. Microbiol. 2013, 51, 1110. [Google Scholar] [CrossRef]
- Escadafal, C.; Faye, O.; Sall, A.A.; Faye, O.; Weidmann, M.; Strohmeier, O.; von Stetten, F.; Drexler, J.; Eberhard, M.; Niedrig, M.; et al. Rapid Molecular Assays for the Detection of Yellow Fever Virus in Low-Resource Settings. PLoS Negl. Trop. Dis. 2014, 8, e2730. [Google Scholar] [CrossRef]
- Kersting, S.; Rausch, V.; Bier, F.F.; von Nickisch-Rosenegk, M. Multiplex isothermal solid-phase recombinase polymerase amplification for the specific and fast DNA-based detection of three bacterial pathogens. Mikrochim. Acta 2014, 181, 1715–1723. [Google Scholar] [CrossRef]
- Teoh, B.T.; Sam, S.S.; Tan, K.K.; Danlami, M.B.; Shu, M.H.; Johari, J.; Hooi, P.S.; Brooks, D.; Piepenburg, O.; Nentwich, O.; et al. Early Detection of Dengue Virus by Use of Reverse Transcription-Recombinase Polymerase Amplification. J. Clin. Microbiol. 2015, 53, 830. [Google Scholar] [CrossRef]
- Zhang, S.; Ravelonandro, M.; Russell, P.; McOwen, N.; Briard, P.; Bohannon, S.; Vrient, A. Rapid diagnostic detection of plum pox virus in Prunus plants by isothermal AmplifyRP((R)) using reverse transcription-recombinase polymerase amplification. J. Virol. Methods 2014, 207, 114–120. [Google Scholar] [CrossRef]
- Tu, P.A.; Shiu, J.S.; Lee, S.H.; Pang, V.F.; Wang, D.C.; Wang, P.H. Development of a recombinase polymerase amplification lateral flow dipstick (RPA-LFD) for the field diagnosis of caprine arthritis-encephalitis virus (CAEV) infection. J. Virol. Methods 2017, 243, 98–104. [Google Scholar] [CrossRef]
- Soliman, H.; Kumar, G.; El-Matbouli, M. Recombinase polymerase amplification assay combined with a lateral flow dipstick for rapid detection of Tetracapsuloides bryosalmonae, the causative agent of proliferative kidney disease in salmonids. Parasites Vectors 2018, 11, 234. [Google Scholar] [CrossRef]
- Tian, A.L.; Elsheikha, H.M.; Zhou, D.H.; Wu, Y.D.; Chen, M.X.; Wang, M.; Chen, D.; Zhang, X.C.; Zhu, X.Q. A novel recombinase polymerase amplification (RPA) assay for the rapid isothermal detection of Neospora caninum in aborted bovine fetuses. Vet. Parasitol. 2018, 258, 24–29. [Google Scholar] [CrossRef]
- Sun, K.; Xing, W.; Yu, X.; Fu, W.; Wang, Y.; Zou, M.; Luo, Z.; Xu, D. Recombinase polymerase amplification combined with a lateral flow dipstick for rapid and visual detection of Schistosoma japonicum. Parasit Vectors 2016, 9, 476. [Google Scholar] [CrossRef]
- Rohrman, B.; Richards-Kortum, R. Inhibition of recombinase polymerase amplification by background DNA: A lateral flow-based method for enriching target DNA. Anal. Chem. 2015, 87, 1963–1967. [Google Scholar] [CrossRef]
- Nejsum, P.; Thamsborg, S.M.; Petersen, H.H.; Kringel, H.; Fredholm, M.; Roepstorff, A. Population dynamics of Ascaris suum in trickle-infected pigs. J. Parasitol. 2009, 95, 1048. [Google Scholar] [CrossRef]
- Ravindran, V.B.; Surapaneni, A.; Crosbie, N.D.; Schmidt, J.; Shahsavari, E.; Haleyur, N.; Soni, S.K.; Ball, A.S. A modified approach to recover and enumerate Ascaris ova in wastewater and sludge. PLoS Negl. Trop. Dis. 2018, 13, 1–6. [Google Scholar] [CrossRef]
- Steinbaum, L.; Kwong, L.H.; Ercumen, A.; Negash, M.S.; Lovely, A.J.; Njenga, S.M.; Boehm, A.B.; Pickering, A.J.; Nelson, K.L. Detecting and enumerating soil-transmitted helminth eggs in soil: New method development and results from field testing in Kenya and Bangladesh. PLoS Negl. Trop. Dis. 2017, 11, e0005522. [Google Scholar] [CrossRef]
- Pilotte, N.; Papaiakovou, M.; Grant, J.R.; Bierwert, L.A.; Llewellyn, S.; McCarthy, J.S.; Williams, S.A. Improved PCR-Based Detection of Soil Transmitted Helminth Infections Using a Next-Generation Sequencing Approach to Assay Design. PLoS Negl. Trop. Dis. 2016, 10, e0004578. [Google Scholar] [CrossRef]
- Verweij, J.J.; Stensvold, C.R. Molecular testing for clinical diagnosis and epidemiological investigations of intestinal parasitic infections. Clin. Microbiol. Rev. 2014, 27, 371–418. [Google Scholar] [CrossRef]
- Jensen, M.A.; Fukushima, M.; Davis, R.W. DMSO and betaine greatly improve amplification of GC-rich constructs in de novo synthesis. PLoS ONE 2010, 5, e11024. [Google Scholar] [CrossRef]
- Pai, N.P.; Vadnais, C.; Denkinger, C.; Engel, N.; Pai, M. Point-of-care testing for infectious diseases: Diversity, complexity, and barriers in low- and middle-income countries. PLoS Med. 2012, 9, e1001306. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Primer | Helminth | Sequence (5’ – 3’) |
|---|---|---|---|
| Recombinase polymerase amplification (RPA) primer screening | Asc718F | A. suum/A. lumbricoides | CTAATCTATGATTCAATATCTCGTTGTAATTT |
| Asc881R | AAATTTTTCATATACATCATTATTGTCACG | ||
| Asc709F | CTTATTTAGCTAATCTATGATTCAATATCTCG | ||
| TS596F | T. suis | GTTATTAACGACCAATGCAGATAAGC | |
| TS764R | GTTCAAAGTATTCAAGTTCAGTGTGTC | ||
| TS510F | CATGCTATGTCGGTGAGGTTTAAAGAA |
| Assay | Primer/Probe | Sequence (5’ – 3’) |
|---|---|---|
| RPA-lateral flow (LF) | Asc718F | CTAATCTATGATTCAATATCTCGTTGTAATTT |
| Asc881RB | Biotin-AAATTTTTCATATACATCATTATTGTCACG | |
| Asc767P | FAM-ATGAGCGAGAGAGAATATATACATCAAAACG-Tetrahydrofuran-TCTTAAAAGACGATT-C3 spacer | |
| TS596F | GTTATTAACGACCAATGCAGATAAGC | |
| TS764RD | Digoxigenine-GTTCAAAGTATTCAAGTTCAGTGTGTC | |
| TS661P | FAM-GTGCAGGAACTCTTGAAACATGATGACATT-tetrahydrofuran-CGAACGGCGGATCACTT-C3 spacer |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravindran, V.B.; Khallaf, B.; Surapaneni, A.; Crosbie, N.D.; Soni, S.K.; Ball, A.S. Detection of Helminth Ova in Wastewater Using Recombinase Polymerase Amplification Coupled to Lateral Flow Strips. Water 2020, 12, 691. https://doi.org/10.3390/w12030691
Ravindran VB, Khallaf B, Surapaneni A, Crosbie ND, Soni SK, Ball AS. Detection of Helminth Ova in Wastewater Using Recombinase Polymerase Amplification Coupled to Lateral Flow Strips. Water. 2020; 12(3):691. https://doi.org/10.3390/w12030691
Chicago/Turabian StyleRavindran, Vivek B., Basma Khallaf, Aravind Surapaneni, Nicholas D. Crosbie, Sarvesh K. Soni, and Andrew S. Ball. 2020. "Detection of Helminth Ova in Wastewater Using Recombinase Polymerase Amplification Coupled to Lateral Flow Strips" Water 12, no. 3: 691. https://doi.org/10.3390/w12030691
APA StyleRavindran, V. B., Khallaf, B., Surapaneni, A., Crosbie, N. D., Soni, S. K., & Ball, A. S. (2020). Detection of Helminth Ova in Wastewater Using Recombinase Polymerase Amplification Coupled to Lateral Flow Strips. Water, 12(3), 691. https://doi.org/10.3390/w12030691