1. Introduction
Glyphosate (
N-(PhosphonoMethyl)Glycine) (PMG) is a broad-spectrum systemic herbicide, invented by Franz and brought to market in 1974 by Monsanto under the trade name Roundup [
1,
2].
Glyphosate, absorbed mainly through foliage, inhibits a plant enzyme —5-enolpyruvylshikimate-3-phosphate (EPSP) synthetase (EC 2.5.1.19)—involved in the bio-synthesis of aromatic amino acids (AAA) substrates in a build-up of plant lignins [
3,
4,
5].
A significant fraction of glyphosate sprayed on crops returns to the soil, and in spite of strong adsorption to soil solids [
6], is able to contaminate surface water (runoff and erosion) [
7,
8,
9].
Animal and epidemiological studies published in recent decades point to a potential glyphosate toxicity [
10,
11,
12]. Further, the World Health Organization’s International Agency for Research on Cancer concluded that glyphosate is “probably carcinogenic to humans” [
13] and Anifandis et al. [
14,
15] demonstrated that glyphosate/PMG induce DNA fragmentation.
Therefore, various treatment processes have been investigated to reduce the pesticide concentration in water and to minimize the potential health risks associated with exposure to these chemicals by the consumption of contaminated water [
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26]. Advanced oxidation processes (AOPs) are a key technology to solve pesticide contamination problems during both water and wastewater treatment (
Table 1).
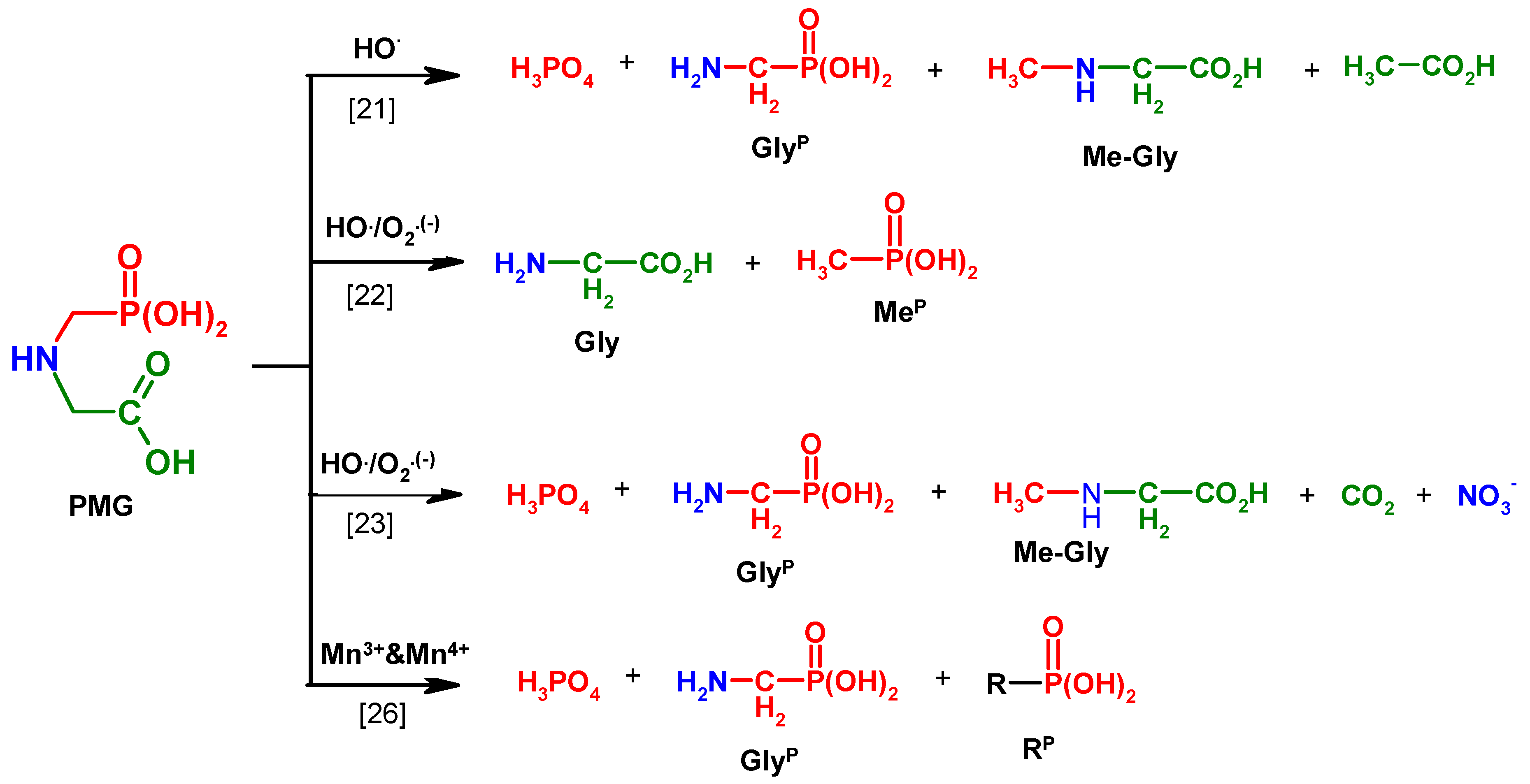
The various mechanisms of PMG degradation using AOP technology, according to the literature, were presented by Manassero [
25]. These not always coherent results (
Figure 1) led us to investigate these process by using
31P NMR monitoring of the PMG degradation processes. This
31P NMR technique has been applied for the analysis of PMG metabolites and degradation products in only a few earlier research works [
26,
27,
28,
29,
30,
31,
32,
33].
2. Materials and Methods
2.1. Materials
The phosphonic amino acids used in the studies (
Table 2) were synthesized as follows: aminomethylphosphonic acid (Gly
P) was synthesized according to the Soroka method [
34,
35]; (
N-methylamino)methylphosphonic acid (Me-Gly
P) was obtained by the hydrophosphonylation of 1,3,5-trimethylhexahydro-1,3,5-triazine by means of diisopropylphosphite according to Maier [
36]; (
N,N-dimethylamino)-methyl-phosphonic acid (Me
2-Gly
P) was obtained by the modified Kabachnik-Fields condensation [
37,
38].
Phosphonomethyl glycine, methylphosphonic acid, 1,3,5-trimethylhexahydro-1,3,5-triazine, catalase and other applied reagents and solvents were purchased from Aldrich. Diisopropylphosphite was purchased from ACROS OrganicTM.
Herbicide Roundup Ultra 170 SL, containing glyphosate-isopropylammonium salt [227.2] (CAS: 38641-94-0; 170 g/L; 15.67%; 0.75 M), and surfactant (CAS not given; 8%) were purchased from Monsanto Europe S.A (Scotts Poland, Warsaw, Poland).
2.2. Synthesis of Aminophosphonic Standards
2.2.1. Synthesis of Aminomethylphosphopnic Acid (GlyP)
Phosphorus chloride(III) (8.75 mL; 0.10 mol) was added dropwise to a well-stirred mixture of
N-(hydroxymethyl)benzamide (synthesized by the hydroxymethylation of benzamide [
34]) (15.1 g; 0.10 mol) and anhydrous acetic acid (20 mL), at ambient temperature. The mixture was then refluxed for 1 h, evaporated (25 °C at 10–20 mm Hg for 15 min, and 75 °C at 0.05 mm Hg) to an oily residue and dissolved in hydrochloric acid solution (5 M aq.; 100 mL). The mixture was heated under reflux for 8 h, cooled to room temperature, and the separated benzoic acid was filtered off. The filtrate was evaporated, the residue was dissolved in water (20 mL), then the solution of crude Gly
P was purified on a Dowex 50 W × 8 ion exchange column using water elution. The obtained Gly
P (8.1 g; 0.072 mol; 72%) was homogeneous at
31P NMR solutions. Elemental analysis data (determined %/(calculated %)) for CH
6NPO
3 [111.04] C = 10.70 (10.82); H = 5.47 (5.45); N = 12.50 (12.61).
2.2.2. Synthesis of N-Methylaminomethylphosphopnic Acid (Me-GlyP)
To trimethylhexahydro-s-triazine (1.29 g; 0.01 mol diisopropylphosphite (5.0 g; 0.03 mol), was added and the mixture was heated with stirring to 100–110 °C for 4 h. The reaction mixture was evaporated (25 °C at 10–20 mm Hg for 15 min, and 75 °C at 0.05 mm Hg), then diluted with 5 M HCl (100 mL) and refluxed for 8 h. The hydrolyzate was evaporated to an oily residue, which was dissolved in water (20 mL) and extracted with ethyl ether (20 mL). The aqueous layer was passed through a Dowex 50 W × 8 ion exchange column using water elution. Fractions were collected (molybdate test) and evaporated. The crystalline product was washed with ethanol, filtered, and dried to give 2.0 g (53.3%) of Me-GlyP, homogeneous 31P NMR solutions. Elemental analysis data (determined %/(calculated %)) for C2H8NPO3 [125.06]: C = 19.12 (19.21); H = 6.55 (6.45); N = 11.10 (11.2).
2.2.3. Synthesis of N,N-Dimethylaminomethylphosphopnic Acid (Me2-GlyP)
The formaldehyde aqueous solution (37%; d = 1.11 g/mL; 2.9 g; 0.05 mol) was gradually added to a stirred mixture of equimolar quantities of dimethylamine (2 M solution of Me2NH in methanol; 25 mL; 0.05 mol) and diethyl phosphite (7.8 g; 0.05 mol), keeping the temperature below 85 °C. The reaction mixture was stirred for 30 min, and evaporated (25 °C at 10–20 mm Hg for 15 min, and 75 °C at 0.05 mm Hg) to an oily residue. The residue was dissolved in 5 M HCl (100 mL) and the solution was refluxed for 8 h. The hydrolyzate was evaporated to dryness (60 °C; 10–20 mm), and the residue was passed through a Dowex 50 W × 8 ion exchange column using water elution. The collected fractions (phosphomolybdate assay) were evaporated to dryness giving white crystals of Me2GlyP (4.2 g; 60.0%), homogeneous in31P NMR solutions. Elemental analysis data (determined %/(calculated %)) for C3H10NPO3 [139.09]: C = 25.78 (25.91); H = 7.32 (7.25); N = 9.98 (10.07).
2.2.4. Solutions
Solution of 0.01 M Fe(II): a sample of FeSO4 × 7H2O [278] (28 mg) was dissolved in water (10 mL).
Solution of 0.02 M Fe(II): a sample of FeSO4 × 7H2O [278] (56 mg) was dissolved in water (10 mL).
Catalase solution: a sample of 10 mg of catalase was dissolved in 50 mL of distilled or deionized water.
Solution of 2 M H2SO4 (in 20% D2O): samples of 2.5 M H2SO4 (20 mL) were diluted to 25 ml with D2O (5 mL).
2.3. Instruments
31P NMR spectra were recorded on a Bruker AC 200 spectrometer operating at 81.01 MHz and on a Bruker Avance III 600 spectrometer operating at 242.9 MHz. 1H NMR spectra were recorded on a Bruker Avance III 600 spectrometer operating at 600 Hz. Positive chemical shift values of 31P were reported for compounds absorbed at lower fields than H3PO4.
The pH measurements were performed using a CX-505 multifunction laboratory meter (Elmetron, Zabrze, Poland) equipped with a combination pH electrode EPP-1 (Elmetron, Zabrze, Poland). The chemical degradation of aqueous solutions of the Roundup herbicide formulation in PMG-H
2O
2 and PMG-H
2O
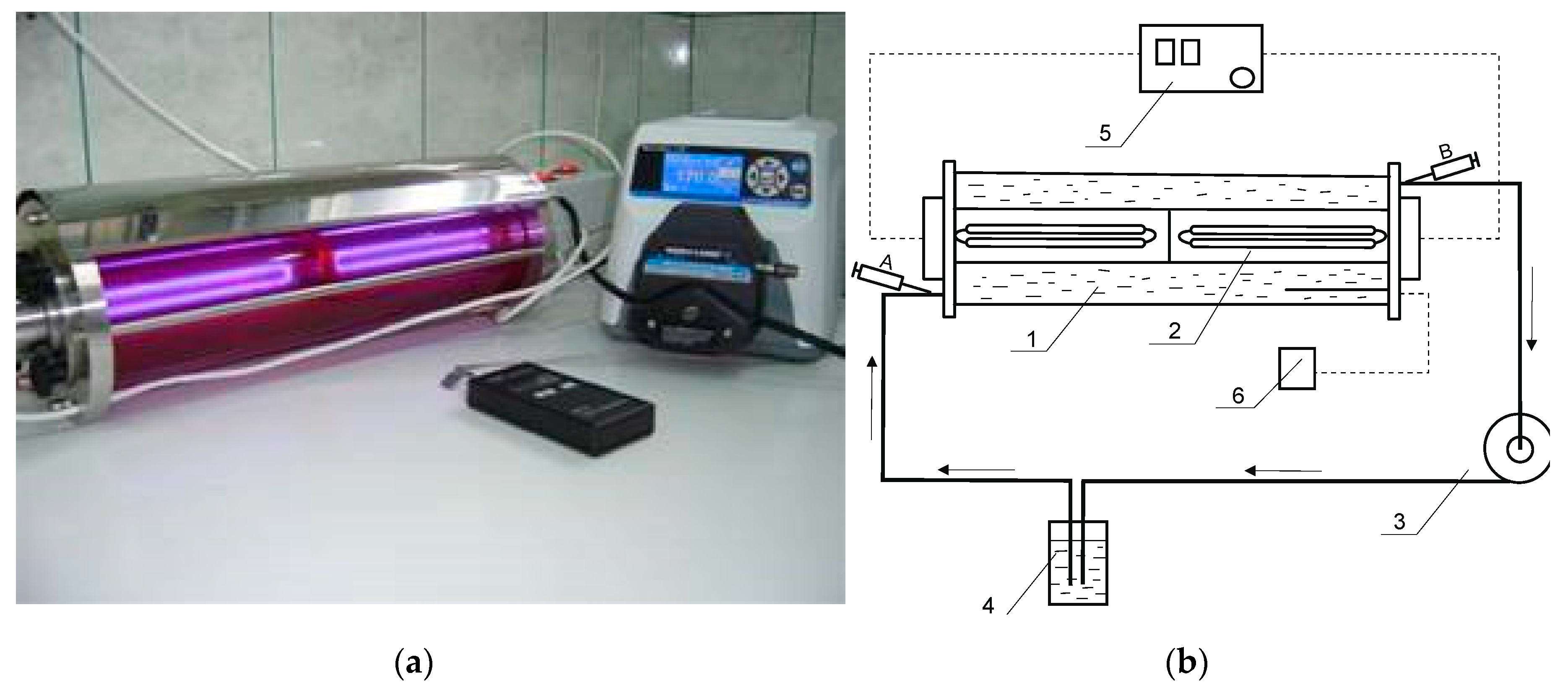
2-UV systems was carried out. Experiments were performed in the reactor shown in
Figure 2.
2.4. Degradation of Glyphosate
2.4.1. Degradation of Phosphonomethyl Glycine (PMG)
Samples of PMG (0.1 mmol) were dissolved in appropriate volumes of FeSO
4 solution (0.5 mL of 0.01 M/0.02 M solution of FeSO
4), followed by the addition of dihydrogen dioxide (the exact amounts of the solutions are given in
Table 3) and kept at room temperature for a desired reaction time. Then, the reaction mixtures were centrifuged, acidified with 2 M H
2SO
4 to pH = 3.5 if necessary, and the volumes of 0.4 mL were taken and mixed with D
2O (0.05 mL) and 0.1 M EDTA (0.05 mL), and analyzed by means of
31P NMR.
2.4.2. Degradation of Roundup Herbicide Formulation by Means of AOP Technology
Reaction mixtures were prepared in accordance with
Table 4. Thus, samples of PMG contained in Roundup Ultra 170 SL Herbicide solution (0.75 M; 16 mL; 12 mmol of PMG), were diluted in water (3680 mL), dihydrogen dioxide samples (60 mmol) were added, and the reaction mixtures were adjusted to the desired pH value by means of acidification with 2 M H
2SO
4 or alkalized by means of 5 M KOH. The oxidative degradations of herbicide were performed for the desired time, during which the reaction progress was monitored by the
31P NMR analysis. Thus, the appropriate samples (4 mL) were treated with catalase (0.1 mL), kept for 30 min at room temperature, and evaporated to an oily residue. These were dissolved in 2 M H
2SO
4 (in 20% D
2O) (0.5 mL) and analyzed using
31P NMR.
3. Results and Discussion
3.1. Protonation Equilibria of Reagents
Representative values of pK
a are given in
Table 5. On this basis, protonation equilibria of phosphonomethyl glycine (PMG) are presented in
Figure 3.
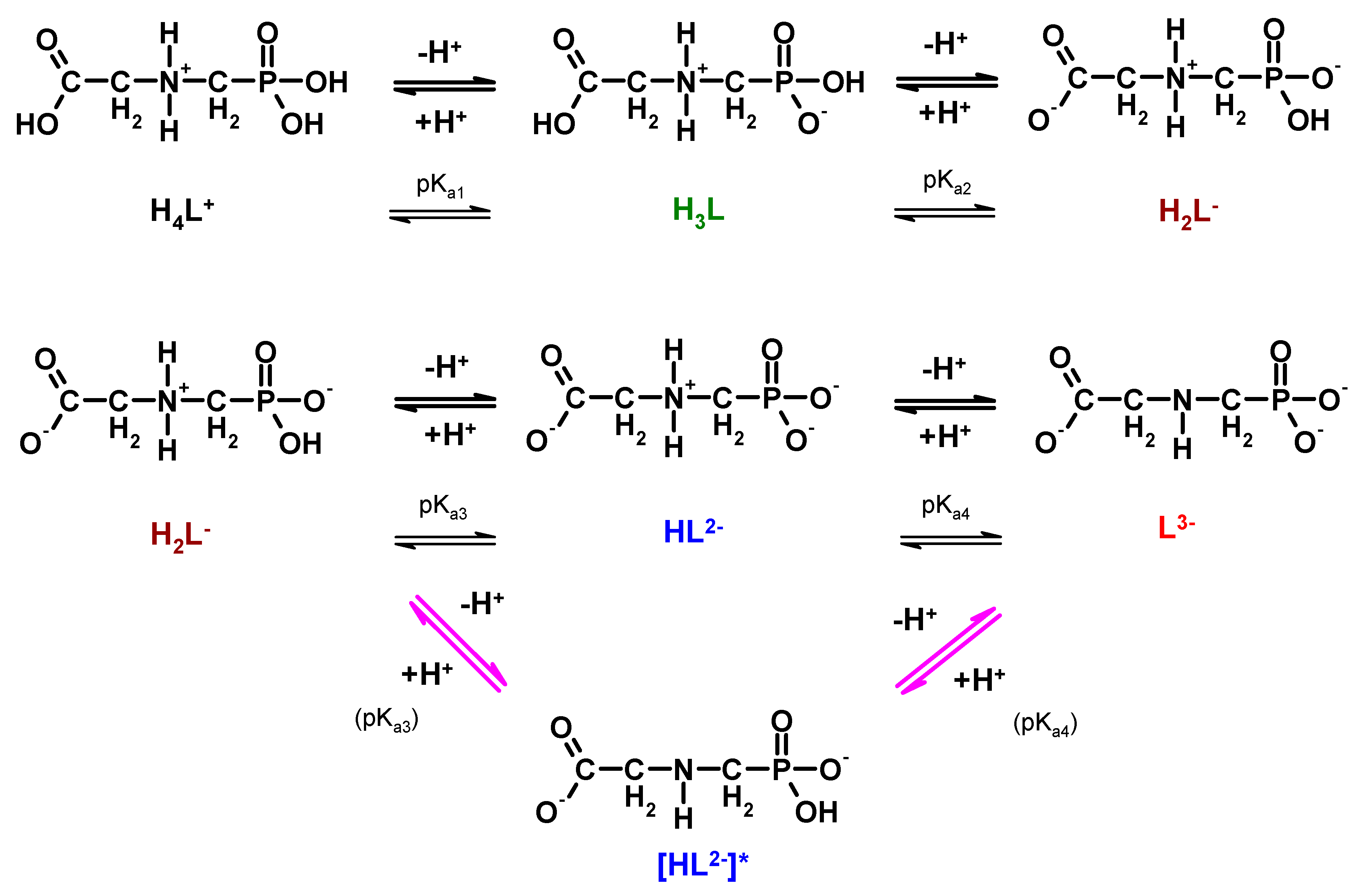
Despite the large amount of research data, there is conflicting information in the literature concerning the first protonation step of glyphosate, namely whether the first dissociable proton in the HL
2− formations is attached to the nitrogen atom of the amino group or to the oxide atom of the phosphonate function. As a matter of fact, only two recent reports consider that the first protonation step occurs on the one of the oxygen atoms in the phosphonate group (
Figure 4, magenta arrows) [
45,
46].
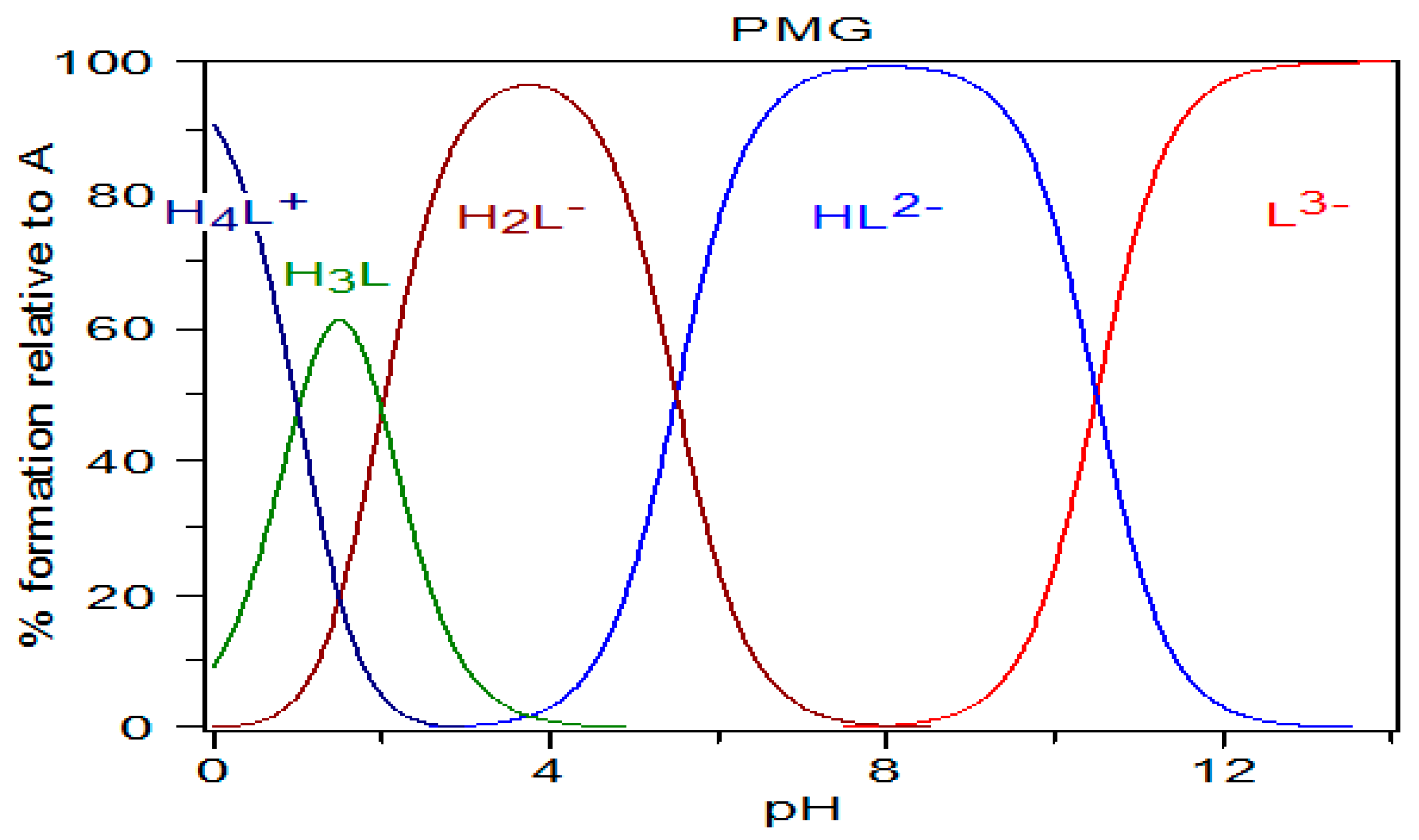
On the basis of this analysis of the speciation graph (
Figure 4), we assumed that in aqueous solutions PMG exists in the following forms: at pH = 0—mainly as H
4L
+ form; at pH = 1.5—mainly as H
3L form; at pH = 3.5–4—mainly as H
2L
−; at pH = 8—mainly as HL
2−; and at pH ≥ 12—mainly as L
3− form.
The protonation equilibrium of dihydrogen dioxide is shown in
Figure 5; dihydrogen dioxide speciation is also presented in
Figure 6. In the literature, the pKa values for H
2O
2 are as follows: pK
a1 = −3.1 [
48] and pK
a2 = 11.6 [
49]. This means that in concentrated H
2SO
4 (e.g., 2 M), dihydrogen dioxide can exist in the H
3L
+ form, at pH = 0–8 it exists in the molecular form H
2L, at pH = 14 it is dissociated in ca. 50% to HL
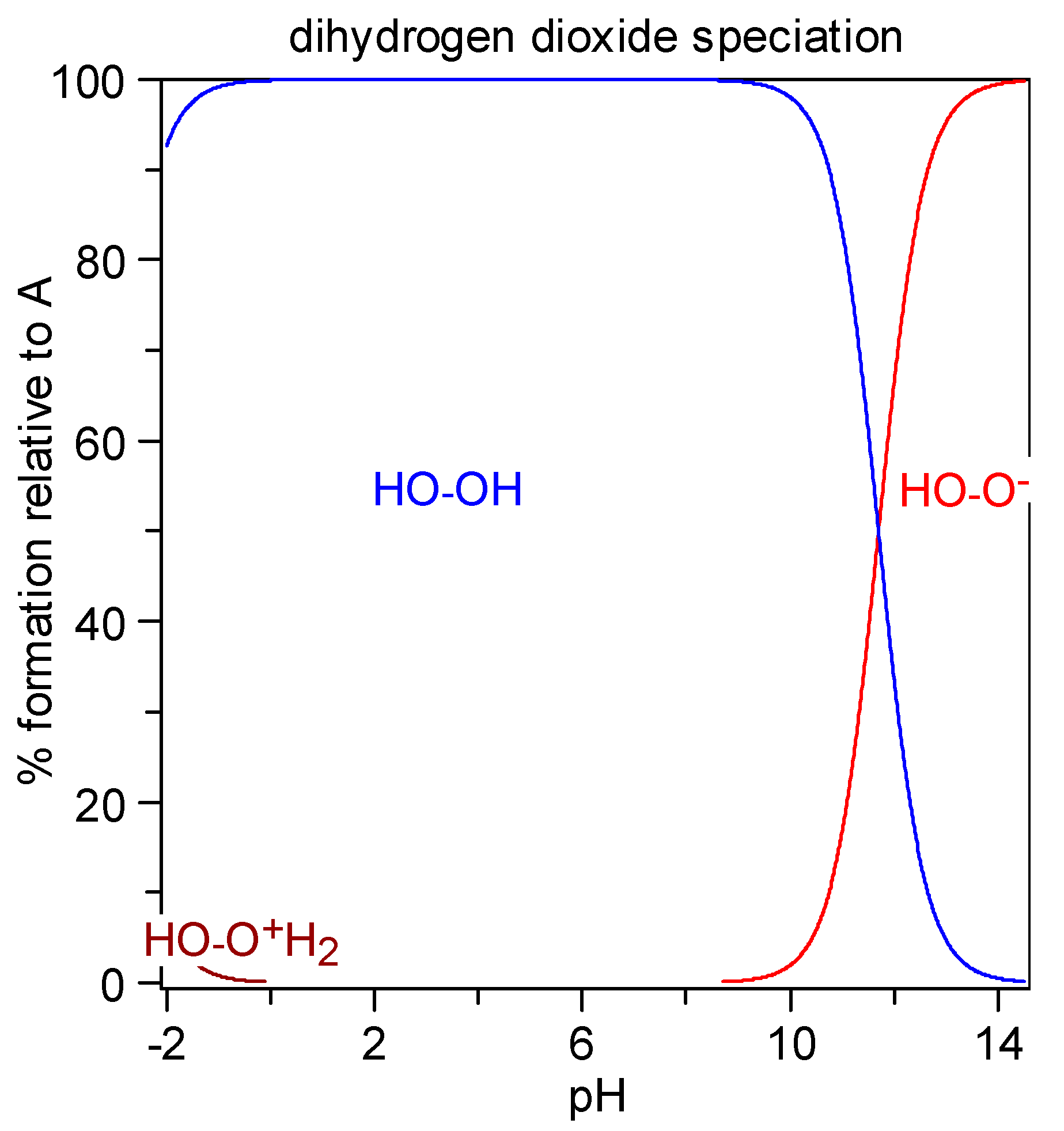
−, and for 2 M KOH (pH > 14) it is almost fully ionized.
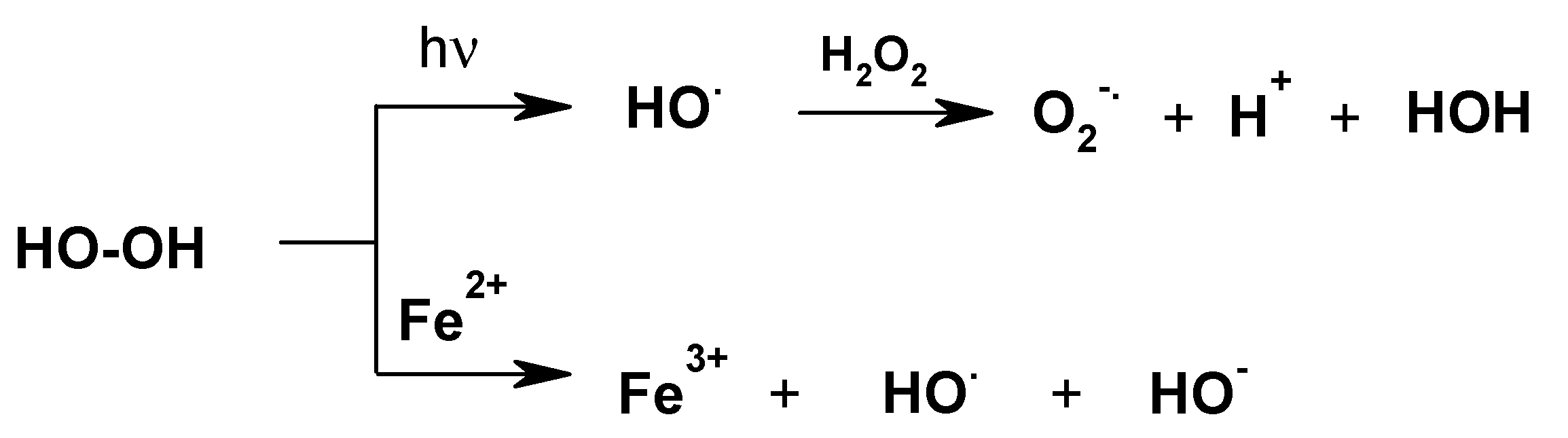
3.2. Reaction of PMG and H2O2
It is generally known that the oxidation potential of H
2O
2 greatly increases during UV irradiation (Mierzwa et al., 2018, and references cited therein) [
50] as well as in the presence of metal ions (
Figure 7) [
50,
51,
52,
53,
54,
55].
Therefore, we assumed that the reaction between PMG and dihydrogen peroxide consumed either the molecular form of H
2O
2 in the absence of irradiation, or hydroxide and peroxide radicals during UV irradiation or in the presence of Fenton reagents. The results of PMG degradation in both modes are illustrated in
Figure 8.
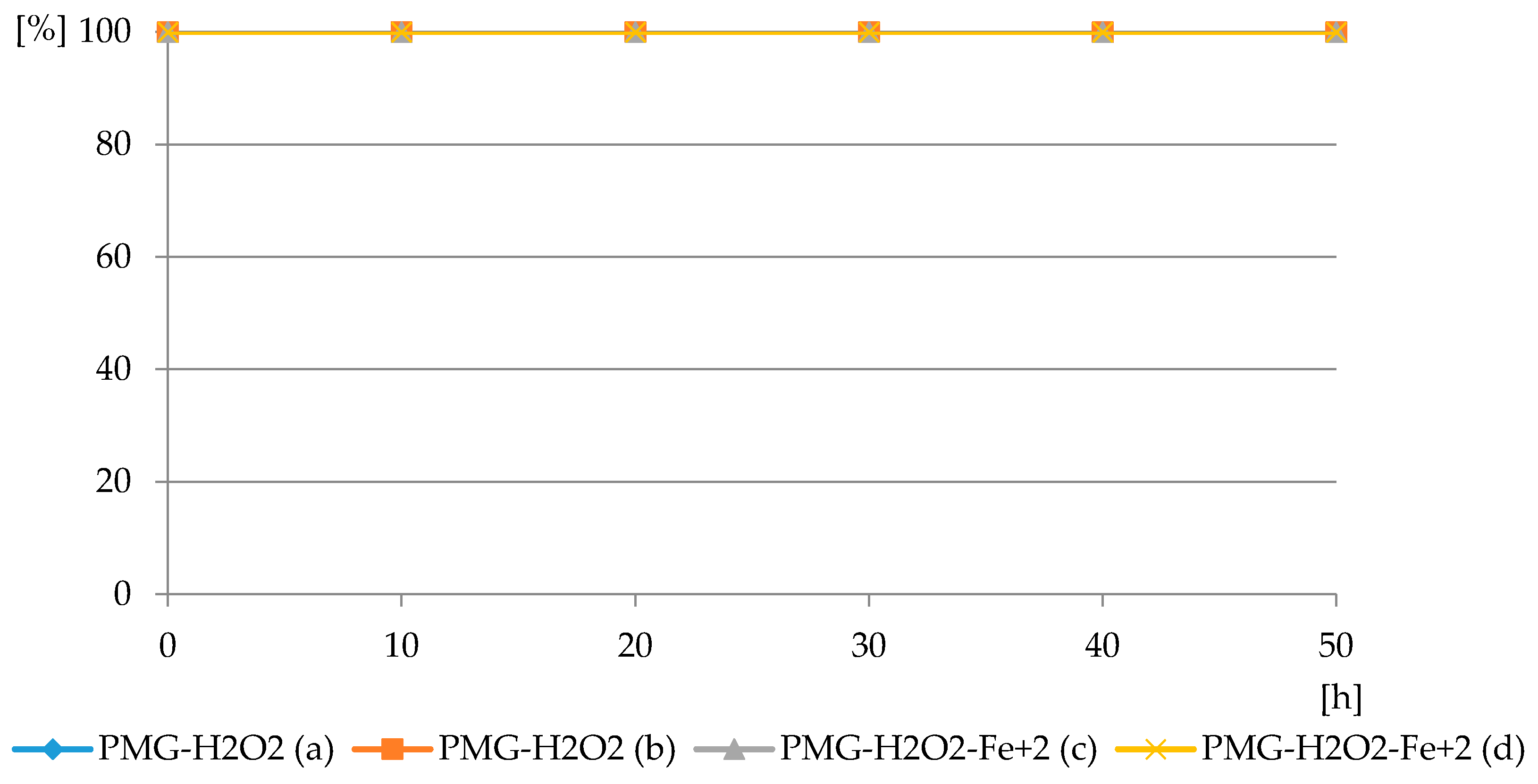
The reaction of PMG with H
2O
2, both with H
2O
2 and H
2O
2-Fe
3+, did not occur at the applied pH = 3.5, at which PMG exists mainly in the H
2L
− protonated on nitrogen form (
Figure 3 and
Figure 4) and hydrogen peroxide mainly in H
2L forms (
Figure 5 and
Figure 6). Therefore, we assumed that the protonation of the amino function in PMG efficiently reduces the interaction of PMG and H
2O
2 (no trace of P-C bond splitting was observed in a 48-h period) (
Figure 8). However, during the irradiation of aqueous solutions of PMG (in the form of the herbicide Roundup) and H
2O
2 (1:5), for a pH range of 2 ≤ pH ≤ 12, the splitting of the P-C bond of PMG was observed, to an extent dependent on the pH of the applied solution (
Figure 9 and
Figure 10). Is worth noting that the irradiation of aqueous PMG solution without H
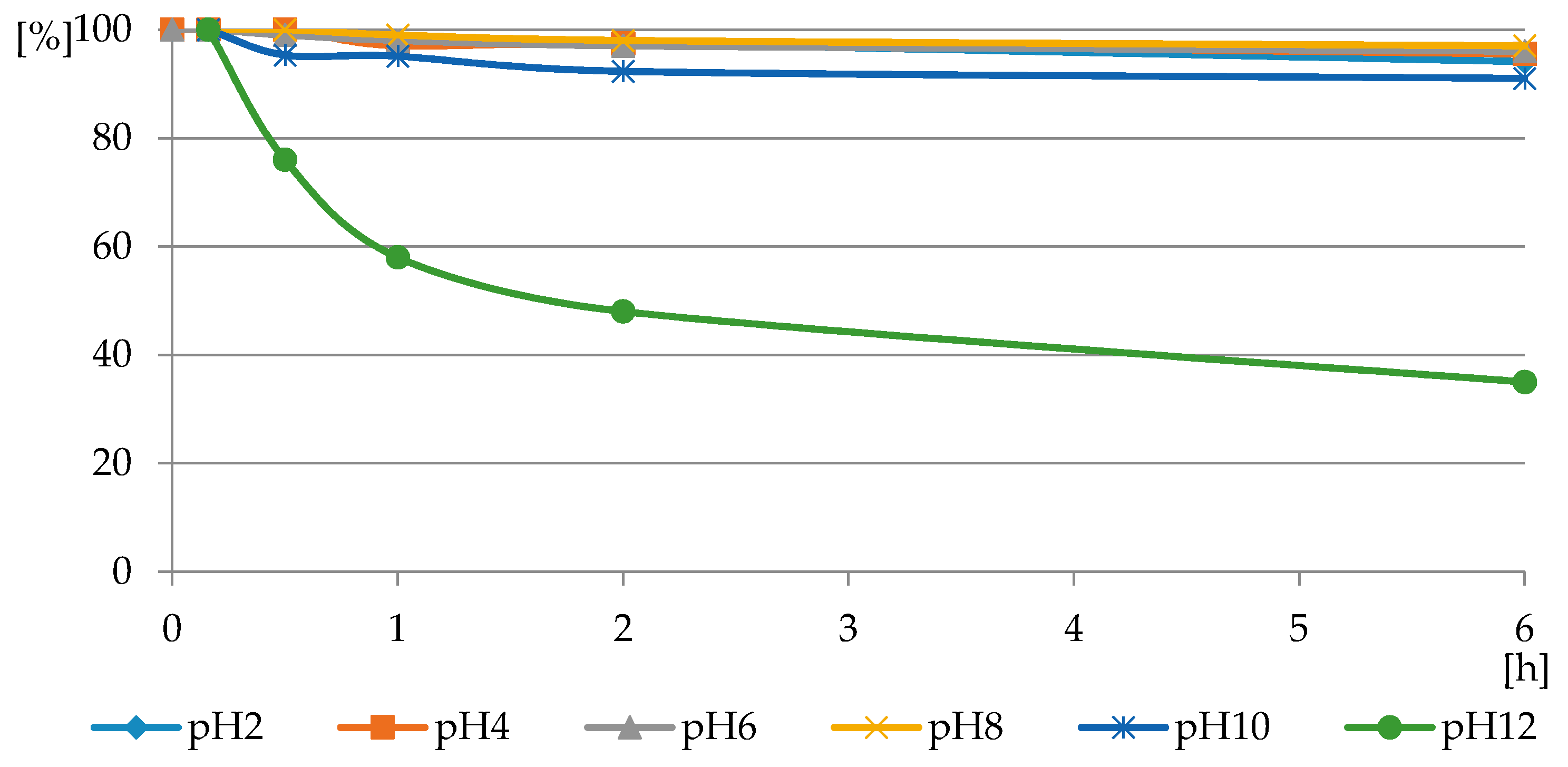
2O
2 during a 48-h period did not exhibit any sign of PMG decomposition (100% of PMG).
The residual PMG quantities were calculated from the corresponding
31P NMR spectra using Equation (1):
where S
(PMG), S
(R−P), and S
(Pi) are the
31P signals corresponding to PMG, phosphonic acids, and inorganic phosphate, respectively.
The
31P NMR spectra of the degradation mixtures of PMG-H
2O
2-(UV) (UV; 25 °C) recorded for reactions carried out at pH = 2, 8, 10, and 12 are presented in
Figure 11. For the identification of the reaction products of PMG-H
2O
2-(UV) mixtures, we recorded the
31P NMR spectra of PMG potential degradation products. The chemical shifts (δ(
31P)) of these compounds are listed in
Table 6.
The results of
31P NMR investigations on PMG degradation with H
2O
2 are shown graphically in
Figure 12.
4. Conclusions
The data presented suggest the PMG inertness toward H2O2 in the modes without UV irradiation, both with (PMG-H2O2-Fe2+) as well as without Fe2+ catalyst (PMG-H2O2). The data considering the reaction modes of PMG with H2O2 under UV irradiation (PMG- H2O2-(UV)) exhibit the slow degradation of PMG at 2 ≤ pH ≤ 10, which becomes faster at pH = 12. The analysis of the 31P NMR spectra of PMG-H2O2 reaction mixtures obtained for reactions carried out at 2 ≤ pH ≤ 10 indicate the presence of initial PMG and H3PO4, and the mixture of PMG, GlyP, and H3PO4/HxPO43−x for reactions carried out at pH = 12. The results suggest the slow formation of an intermediate PMG × H2O2 phase in the first stage of degradation which decomposes very fast (no intermediates were observed in the 31P NMR spectra) by the scission of the P-C bond of PMG and the subsequent release of phosphoric acid/phosphate ion.
Recapitulating, in the experiments carried out without UV radiation we observed:
In the experiments carried out with UV radiation (PMG-H2O2-(UV)), the P-C rapture type of PMG degradation was observed, the extent of which was dependent on the applied pH of the reaction mixtures. As a result, for the reactions run at 2 ≤ pH ≤ 10, the partial formation of phosphoric acid/phosphate ions (PMG → PMG + H3PO4/HxPO43−x) was observed, whereas for reactions run at pH = 12, mixtures of PMG, GlyP, and PO43− were found.
We did not observe:
Author Contributions
M.H.K. designed the research study and contributed to the data interpretation and to the manuscript drafting and revisions and was involved in the concept of the research study, analyzed the data, and contributed to writing the manuscript. R.Ż. participated in the publication preparation. Z.M. performed experiments. P.U. recorded N.M.R. spectra and analyzed the experimental data.
Funding
This research was funded by the Polish Ministry of Science and Higher Education within statutory research work carried out in 2018 at the Textile Research Institute, Łódź, Poland.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Franz, J.E.; Mao, M.K.; Sikorski, J.A. Glyphosate: A Unique Global Herbicide; American Chemical Society: Washington, DC, USA, 1997; ISBN 0841234582. [Google Scholar]
- Dill, G.M.; Sammons, R.D.; Feng, P.C.C.; Kohn, F.; Kretzmer, K.; Mehrsheikh, A.; Bleeke, M.; Honegger, J.L.; Farmer, D.; Wright, D.; et al. Glyphosate: discovery, development, applications, and properties. In Glyphosate Resistance in Crops and Weeds. History, Development and Management; Nandula, V.K., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; ISBN 978-0-470-41031-8. [Google Scholar]
- Amrhein, N.; Deus, B.; Gehrke, P.; Steinrucken, H.C. The site of the inhibition of the shikimate pathway by glyphosate. II. Interference of glyphosate with chorismate formation in vivo and in vitro. Plant Physiol. 1980, 66, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, J.A.; Gruys, K.J. Understanding glyphosate’s molecular mode of action with EPSP synthase: Evidence favoring an allosteric inhibitor model. Acc. Chem. Res. 1997, 30, 2–8. [Google Scholar] [CrossRef]
- Sviridov, A.V.; Shushkova, T.V.; Ermakova, I.T.; Ivanova, E.V.; Leontievsky, A.A. Glyphosate: Safety Risks, Biodegradation, and Bioremediation. In Current Environmental Issues and Challenges; Cao, G., Orru, R., Eds.; Springer Science+Business Media: Dordrecht, The Netherlands, 2014; pp. 183–195. [Google Scholar] [CrossRef]
- Rampazzo, N.; Todorovic, G.; Mentler, A.; Blum, W.E.H. Adsorption of glyphosate and aminomethylphosphonic acid in soils. Int. Agrophys. 2013, 27, 203–209. [Google Scholar] [CrossRef]
- Borggaard, O.K.; Gimsing, A.L. Fate of glyphosate in soil and the possibility of leaching to ground and surface waters: A review. Pest. Manag. Sci. 2008, 64, 441–456. [Google Scholar] [CrossRef]
- Aparicio, V.C.; De Gerónimo, E.; Marino, D.; Primost, J.; Carriquiriborde, P.; Costa, J.L. Environmental fate of glyphosate and aminomethylphosphonic acid in surface waters and soil of agricultural basins. Chemosphere 2013, 93, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Mamy, L.; Barriuso, E.; Gabrielle, B. Glyphosate fate in soils when arriving in plant residues. Chemosphere 2016, 154, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.M.; Kroes, R.; Munro, I.C. Safety evaluation and risk assessment of the herbicide Roundup and its active ingredient, glyphosate, for humans. Regul. Toxicol. Pharmacol. 2000, 31, 117–165. [Google Scholar] [CrossRef]
- Mesnage, R.; Defarge, N.; Vendomois, J.S.; Seralini, G.E. Potential toxic effects of glyphosate and its commercial formulations below regulatory limits. Food Chem. Toxicol. 2015, 84, 133–153. [Google Scholar] [CrossRef]
- Myers, J.P.; Antoniou, M.N.; Blumberg, B.; Caroll, L.; Colborn, T.; Everett, L.G.; Hansen, M.; Landrigan, P.J.; Lanphear, B.P.; Mesnage, R.; et al. Concerns over use of glyphosate-based herbicides and risks associated with exposures: A consensus statement. Environ. Health 2016, 15, 19. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Loomis, D.; Grosse, Y.; Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Scoccianti, C.; Mattock, H.; Straif, K. Carcinogenicity of tetrachlorvinphos, parathion, malathion, diazinon, and glyphosate. Lancet Oncol. 2015, 16, 490–491. [Google Scholar] [CrossRef]
- Anifandis, G.; Katsanaki, K.; Lagodonti, G.; Messini, C.; Simopoulou, M.; Dafopoulos, K.; Daponte, A. The effect of glyphosate on human sperm motility and sperm DNA fragmentation. Int. J. Environ. Res. Public Health 2018, 15, 1117. [Google Scholar] [CrossRef] [PubMed]
- Anifandis, G.; Amiridis, G.; Dafopoulos, K.; Daponte, A.; Dovolou, E.; Gavriil, E.; Gorgogietas, V.; Kachpani, E.; Mamuris, Z.; Messini, C.I.; et al. The in vitro impact of the herbicide roundup on human sperm motility and sperm mitochondria. Toxics 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Rueppel, M.L.; Brightwell, B.B.; Schaefer, R.; Marvel, J.T. Metabolism and degradation of glyphosate in soil and water. J. Agric. Food Chem. 1977, 25, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Gohre, K.; Casida, J.E.; Ruzo, L.O. N-oxidation and cleavage of the amino acid derived herbicide glyphosate. J. Agric. Food Chem. 1987, 35, 388–392. [Google Scholar] [CrossRef]
- Ikehata, H.; Gama El-Din, M. Aqueous pesticide degradation by hydrogen peroxide/ultraviolet irradiation and Fenton-type advanced oxidation processes: A review. J. Environ. Eng. Sci. 2006, 5, 81–135. [Google Scholar] [CrossRef]
- Jaisi, D.P.; Li, H.; Wallace, A.F.; Paudel, P.; Sun, M.; Balakrishna, A.; Lerch, R.N. Mechanisms of bond cleavage during manganese oxide and UV degradation of glyphosate: Results from phosphate oxygen isotopes and molecular simulations. J. Agric. Food Chem. 2016, 64, 8474–8482. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.A.; McBride, M.B. Oxidative degradation of glyphosate and amino-methylphosphonate by manganese oxide. Environ. Sci. Technol. 2005, 39, 9223–9228. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, F.; Lin, Y.; Deng, N.; Bazhin, N.; Glebov, E. Photodegradation of glyphosate in the ferrioxalate system. J. Hazard. Mater. 2007, 148, 360–365. [Google Scholar] [CrossRef]
- Muneer, M.; Boxall, C. Photocatalyzed degradation of a pesticide derivative glyphosate in aqueous suspensions of titanium dioxide. Int. J. Photoenergy 2008. [Google Scholar] [CrossRef]
- Echavia, G.R.M.; Matzusawa, F.; Negishi, N. Photocatalytic degradation of organophosphate and phosphonoglycine pesticides using TiO2 immobilized on silica gel. Chemosphere 2009, 76, 595–600. [Google Scholar] [CrossRef]
- Assalin, M.R.; De Moraes, S.G.; Queiroz, S.C.; Ferracini, V.; Duran, N. Studies on degradation of glyphosate by several oxidative chemical processes: ozonation, photolysis and heterogeneous photocatalysis. J. Environ. Sci. & Health B 2010, 45, 89–94. [Google Scholar] [CrossRef]
- Manassero, A.; Passalia, C.; Negro, A.C.; Cassano, A.; Zalazar, C.S. Glyphosate degradation in water employing the H2O2/UV-C process. Water Res. 2010, 44, 3875–3882. [Google Scholar] [CrossRef]
- Paudel, P.; Negusse, A.; Jaisi, D.P. Birnessite-catalyzed degradation of glyphosate: a mechanistic study aided by kinetics batch studies and NMR spectroscopy. Soil Sci. Soc. Am. J. 2015, 79, 815–825. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Gralak, D.K.; Drabowicz, J.; Łuczak, J. Novel approach for the simultaneous analysis of glyphosate and its metabolites. J. Chromatogr. A 2002, 947, 129–141. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Gralak, D.K.; Andrijewski, G.; Drabowicz, J.; Łuczak, J. Simultaneous analysis of biologically active aminoalkanephosphonic acids. J. Chromatogr. A 2003, 998, 183–199. [Google Scholar] [CrossRef]
- Cartigny, B.; Azaroual, N.; Imbenotte, M.; Mathieu, D.; Vermeersch, G.; Goulle, J.P.; Lhermitte, M. Determination of glyphosate in biological fluids by 1H and 31P NMR spectroscopy. Forensic. Sci. Int. 2004, 143, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Lipok, J.; Wieczorek, D.; Jewginski, M.; Kafarski, P. Prospects of in vivo 31P NMR method in glyphosate degradation studies in whole cell system. Enzyme Microb. Technol. 2009, 44, 11–16. [Google Scholar] [CrossRef]
- Ge, X.; d’Avignon, D.A.; Ackerman, J.J.H.; Sa, R.D. In vivo 31P-Nuclear Magnetic Resonance studies of glyphosate uptake, vacuolar sequestration, and tonoplast pump activity in glyphosate-resistant horseweed. Plant Physiol. 2014, 166, 1256–1268. [Google Scholar] [CrossRef]
- Cai, H.; Chuang, W.G.; Cui, X.; Cheng, R.H.; Chiu, K.; Chen, Z.; Ding, S. High Resolution 31P NMR Spectroscopy generates a quantitative evolution profle of phosphorous translocation in germinating sesame seed. Sci. Rep. 2018, 8, 359. [Google Scholar] [CrossRef]
- Kudzin, M.H.; Drabowicz, J.; Jordan, F.; Kudzin, Z.H.; Urbaniak, P. Reactivity of aminophosphonic acids. 3. Reaction with hydrogen peroxide. Phosphorus Sulfur Silicon Relat. Elem. 2018, 193. in press. [Google Scholar] [CrossRef]
- Soroka, M. Comments on the synthesis of aminomethylphosphonic acid. Synthesis 1989, 547–548. [Google Scholar] [CrossRef]
- Hellmann, H. New methods of organic preparative chemistry. 8. Amidomethylation. Angew. Chem. 1957, 69, 463–471. [Google Scholar] [CrossRef]
- Maier, L. Organic phosphorus compounds. Part 98. Synthesis of N-methylaminomethylphosphonic acid and derivatives. Phosphorus Sulfur Silicon Relat. Elem. 1991, 62, 29–34. [Google Scholar] [CrossRef]
- Fields, E.K. The Synthesis of esters of substituted amino phosphonic acids. J. Am. Chem. Soc. 1952, 74, 1527–1531. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Kudzin, M.H.; Drabowicz, J.; Stevens, C. Aminophosphonic acids—Phosphorus analogues of natural amino acids. Part 1: Syntheses of α-aminophosphonic acids. Curr. Org. Chem. 2011, 15, 2015–2071. [Google Scholar] [CrossRef]
- Drabowicz, J.; Jakubowski, H.; Kudzin, M.H.; Kudzin, Z.H. The nomenclature of 1-aminoalkylphosphonic acids and derivatives: Evolution of the code system. Acta Biochim. Pol. 2015, 62, 139–150. [Google Scholar] [CrossRef]
- Sprankle, P.; Meggitt, W.F.; Penner, D. Adsorption, mobility, and microbial degradation of glyphosate in the soil. Weed Sci. 1975, 23, 224–234. [Google Scholar] [CrossRef]
- Wauchope, D. Acid Dissociation constants of arsenic acid, methylarsonic acid (MAA), dimethylarsinic acid (cacodylic acid), and N-(Phosphonomethyl)glycine (Glyphosate). J. Agric. Food Chem. 1976, 24, 717–721. [Google Scholar] [CrossRef]
- Appleton, T.; Hall, J.; McMahon, I. NMR spectra of iminobis(methylenephosphonic acid), HN(CH2PO3H2)2, and related ligands and of their complexes with platinum(II). Inorg. Chem. 1986, 25, 726–734. [Google Scholar] [CrossRef]
- Castellino, S.; Leo, G.; Sammons, R.D.; Sikorski, J. 31P, 15N, and 13C NMR of glyphosate: Comparison of pH titrations to the herbicidal dead-end complex with 5-enolpyruvoylshikimate-3-phosphate synthase. Biochemistry 1989, 28, 3856–3868. [Google Scholar] [CrossRef]
- Barja, B.C.; Dos Santos Afonso, M. An ATR−FTIR study of glyphosate and its Fe(III) complex in aqueous solution. Environ. Sci. Technol. 1998, 32, 3331–3335. [Google Scholar] [CrossRef]
- Peixoto, M.M.; Bauerfeldt, G.F.; Herbst, M.H.; Pereira, M.S.; Da Silva, C.O. Study of the stepwise deprotonation reactions of glyphosate and the corresponding pKa values in aqueous solution. J. Phys. Chem. A 2015, 119, 5241–5249. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dong, L.; Yu, Q.; Li, X.; Wu, F.; Tan, Z.; Luo, S. Thermodynamic study on the protonation reactions of glyphosate in aqueous solution: potentiometry, calorimetry and NMR spectroscopy. J. Phys. Chem. B 2016, 120, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): a utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Ermakov, A.N.; Larin, I.K.; Kozlov, Y.N.; Purmal, A.P. The thermodynamic characteristics of hydrogen peroxide in H2SO4-H2O solutions. Russ. J. Phys. Chem. 2006, 80, 1895–1901. [Google Scholar] [CrossRef]
- Kwon, B.G.; Lee, J.H. Determination of hydroperoxyl/superoxide anion radical (HO2/O2−) concentration in the decomposition of ozone using a kinetic method. Bull. Korean Chem. Soc. 2006, 27, 1785–1800. [Google Scholar] [CrossRef]
- Mierzwa, J.C.; Rodrigues, R.; Teixeira, A.C.S.C. UV-Hydrogen Peroxide Processes. In Advanced Oxidation Processes for Wastewater Treatment: Emerging Green Chemical Technology; Chpt. 2; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 13–48. [Google Scholar]
- Aust, S.D.; Morehouse, L.A.; Thomas, C.E. Role of metals in oxygen radical reactions. J. Free Radic. Biol. Med. 1985, 1, 3–25. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. Ser. A 1934, 147, 332–335. [Google Scholar] [CrossRef]
- Weinstein, J.; Bielski, B.H.J. Kinetics of the interaction of HO2 and O2 radicals with hydrogen peroxide. The Haber-Weiss reaction. J. Am. Chem. Soc. 1979, 101, 58–62. [Google Scholar] [CrossRef]
- Zepp, R.G.; Faust, B.C.; Hoigne, J. Hydroxyl radical formation in aqueous reactions (pH 3-8) of iron(II) with hydrogen peroxide: The Photo-Fenton Reaction. Environ. Sci. Technol. 1992, 26, 313–319. [Google Scholar] [CrossRef]
- Duesterberg, C.K.; Mylon, S.E.; Waite, T.D. pH effects on iron-catalyzed oxidation using Fenton’s Reagent. Environ. Sci. Technol. 2008, 42, 8522–8527. [Google Scholar] [CrossRef] [PubMed]
- Rawalay, S.S.; Shechter, H. Oxidation of primary, secondary, and tertiary amines with neutral permanganate. Simple method for degrading amines to aldehydes and ketones. J. Org. Chem. 1967, 32, 3129–3131. [Google Scholar] [CrossRef]
- Mitsui, H.; Zenki, S.I.; Shiota, T.; Murahashi, S.I. Tungstate catalyzed oxidation of secondary amines with hydrogen peroxide. A novel transformation of secondary amines into nitrones. J. Chem. Soc. Chem. Commun. 1984, 874–875. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Drabowicz, J.; Skowroński, R. Synthesis of alkanephosphonates containing nitrone moiety. Phosphorus Sulfur Silicon Relat. Elem. 1990, 51, 310. [Google Scholar] [CrossRef]
- Murahashi, S.I.; Mitsui, H.; Shiota, T.; Tsuda, T.; Watanabe, S. Tungstate-catalyzed oxidation of secondary amines to nitrones. α-Substitution of secondary amines via nitrones. J. Org. Chem. 1990, 55, 1736–1744. [Google Scholar] [CrossRef]
- Colladon, M.; Scarso, A.; Strukul, G. Mild catalytic oxidation of secondary and tertiary amines to nitrones and N-oxides with H2O2 mediated by Pt(II) catalysts. Green Chem. 2008, 10, 793–798. [Google Scholar] [CrossRef]
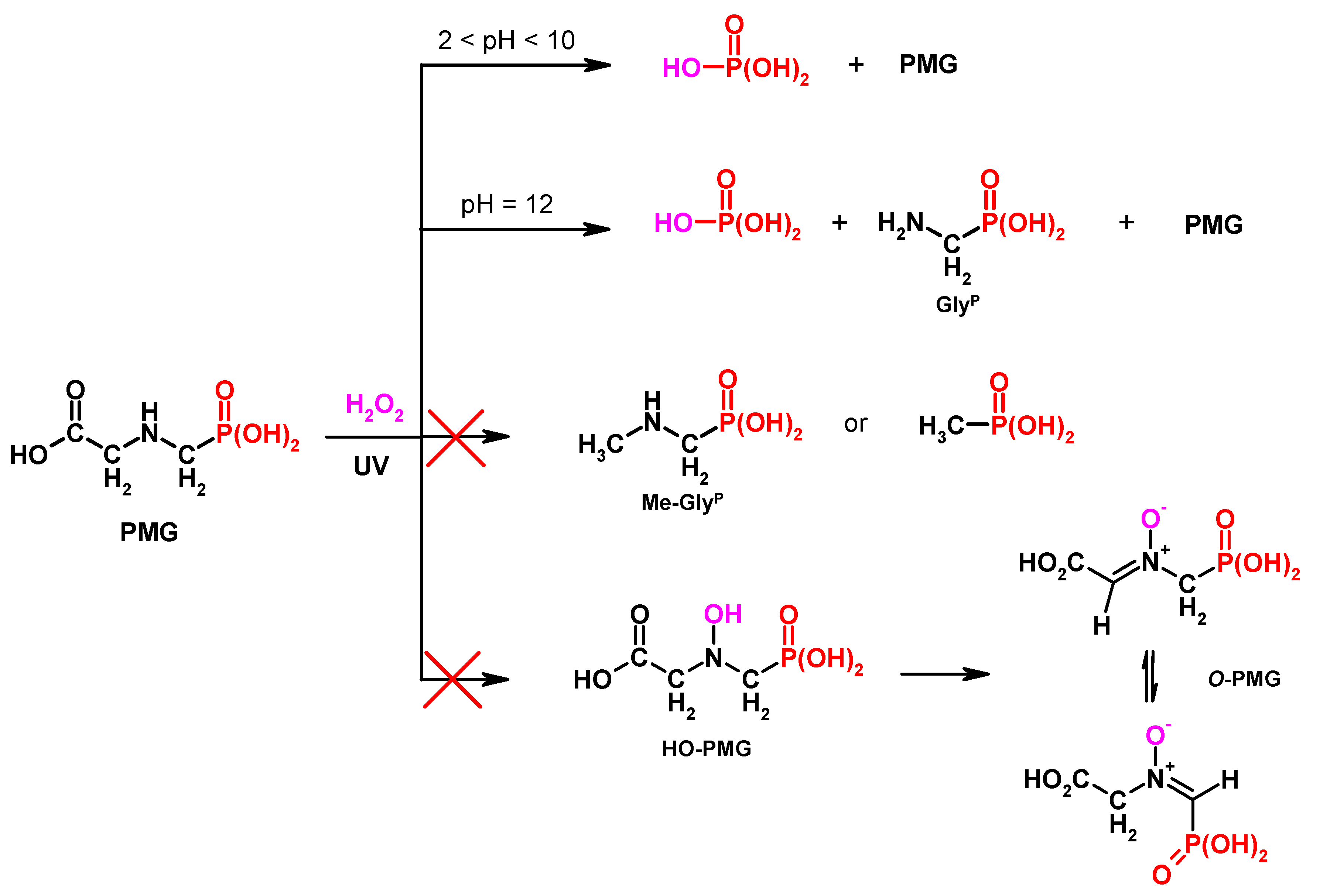
Figure 1.
Degradation products of N-PhosphonoMethyl)Glycine (PMG) of representative methods.
Figure 1.
Degradation products of N-PhosphonoMethyl)Glycine (PMG) of representative methods.
Figure 2.
Reactor used for Roundup degradation by means of AOP technology. (a) Figure of a photoreactor with UV lamp used for the oxidation of PMG by means of UV/H2O2. (b) Schematic diagram: 1—glass reactor; 2—quartz tube with UV lamp (254 nm; 22 W); 3—peristaltic pump; 4—reactor connector; 5—UV power supply; 6—temperature detector; A, B—samples. Applied conditions: PMG (12 mmol), H2O2 (60 mol) in an aq. solution (3.7 L) at different initial pH values applied: 2 ≤ pHs ≤ 12. Irradiation time was up to 360 min. Temperature was 25 °C.
Figure 2.
Reactor used for Roundup degradation by means of AOP technology. (a) Figure of a photoreactor with UV lamp used for the oxidation of PMG by means of UV/H2O2. (b) Schematic diagram: 1—glass reactor; 2—quartz tube with UV lamp (254 nm; 22 W); 3—peristaltic pump; 4—reactor connector; 5—UV power supply; 6—temperature detector; A, B—samples. Applied conditions: PMG (12 mmol), H2O2 (60 mol) in an aq. solution (3.7 L) at different initial pH values applied: 2 ≤ pHs ≤ 12. Irradiation time was up to 360 min. Temperature was 25 °C.
Figure 3.
Dissociation/protonation equilibria of glyphosate (lower branch (arrows in magenta) considering the reports of Peixoto et al., 2015 [
45] and Liu et al., 2016 [
46]).
Figure 3.
Dissociation/protonation equilibria of glyphosate (lower branch (arrows in magenta) considering the reports of Peixoto et al., 2015 [
45] and Liu et al., 2016 [
46]).
Figure 4.
Diagram of PMG species distribution calculated using the pK
a values of Appleton et al. [
42] (pK
a1 < 1; pK
a2 = 2.0; pK
a3 = 5.5; pK
a4 = 10.5), and the HySS program (Alderighi et al., 1999) [
47].
Figure 4.
Diagram of PMG species distribution calculated using the pK
a values of Appleton et al. [
42] (pK
a1 < 1; pK
a2 = 2.0; pK
a3 = 5.5; pK
a4 = 10.5), and the HySS program (Alderighi et al., 1999) [
47].
Figure 5.
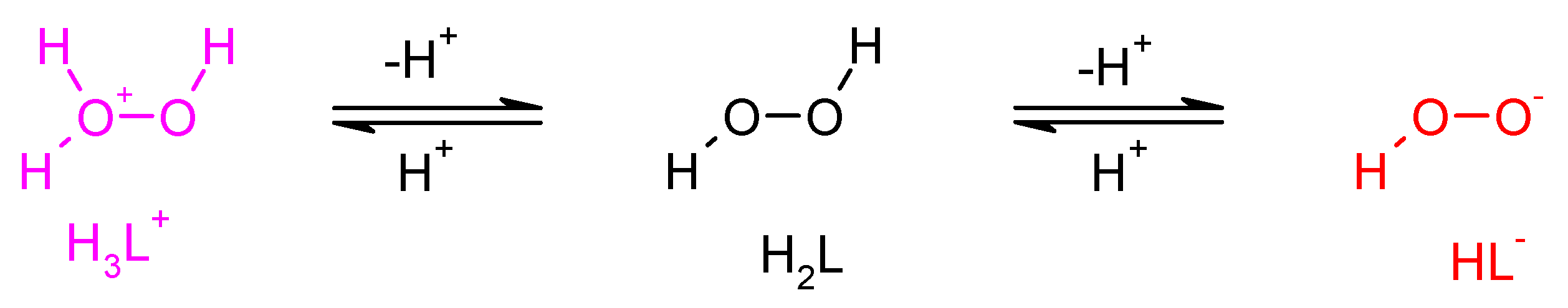
Dissociation/protonation equilibria of dihydrogen dioxide.
Figure 5.
Dissociation/protonation equilibria of dihydrogen dioxide.
Figure 6.
Diagram of dihydrogen dioxide distribution: percent species formation calculated with the HySS program (Alderighi et al., 1999) [
47] for a 10 millimolar solution of H
2O
2 (pK
a1 = −3.1, pK
a2 = 11.7).
Figure 6.
Diagram of dihydrogen dioxide distribution: percent species formation calculated with the HySS program (Alderighi et al., 1999) [
47] for a 10 millimolar solution of H
2O
2 (pK
a1 = −3.1, pK
a2 = 11.7).
Figure 7.
Activation of dihydrogen peroxide via the generation of radicals.
Figure 7.
Activation of dihydrogen peroxide via the generation of radicals.
Figure 8.
The profiles of PMG consumption in reaction with H2O2 in PMG-H2O2 and PMG- H2O2-Fe2+ systems at pH = 3.5: PMG:H2O2 = 1:5 (PMG-H2O2 (a)) and 1:10 (PMG-H2O2 (b)); PMG:H2O2:Fe2+ = 1:10:0.05 (PMG-H2O2-Fe2+ (c)); and 1:10:0.1 (PMG-H2O2-Fe2+ (d)) (residual PMG (%) vs. exposure time (h)).
Figure 8.
The profiles of PMG consumption in reaction with H2O2 in PMG-H2O2 and PMG- H2O2-Fe2+ systems at pH = 3.5: PMG:H2O2 = 1:5 (PMG-H2O2 (a)) and 1:10 (PMG-H2O2 (b)); PMG:H2O2:Fe2+ = 1:10:0.05 (PMG-H2O2-Fe2+ (c)); and 1:10:0.1 (PMG-H2O2-Fe2+ (d)) (residual PMG (%) vs. exposure time (h)).
Figure 9.
The profile of PMG consumption in reactions with H2O2 in PMG-H2O2-(UV) systems (with UV irradiation) carried out at a pH range of 2 ≤ pH ≤ 12 and a temperature of 25 °C (residual PMG (%) vs. exposure time (h)).
Figure 9.
The profile of PMG consumption in reactions with H2O2 in PMG-H2O2-(UV) systems (with UV irradiation) carried out at a pH range of 2 ≤ pH ≤ 12 and a temperature of 25 °C (residual PMG (%) vs. exposure time (h)).
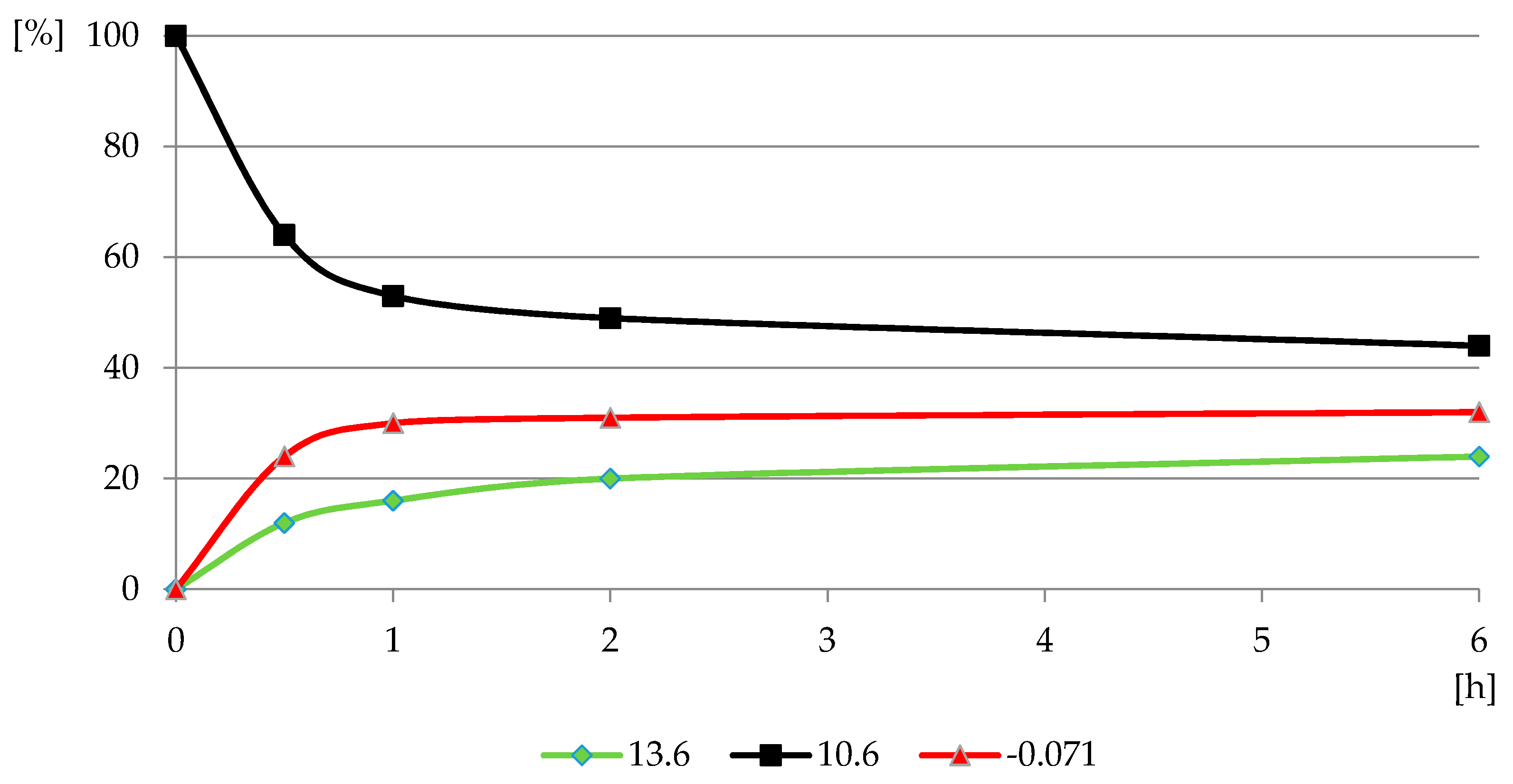
Figure 10.
The profile of PMG reaction with H2O2 in PMG-H2O2-(UV) systems (UV; 25 °C) carried out at pH = 12 (relative P-compounds contribution (GlyP: 13.6 ppm, PMG: 10.6 ppm; Pi: −0.071 ppm) (%) vs. exposition time (h)).
Figure 10.
The profile of PMG reaction with H2O2 in PMG-H2O2-(UV) systems (UV; 25 °C) carried out at pH = 12 (relative P-compounds contribution (GlyP: 13.6 ppm, PMG: 10.6 ppm; Pi: −0.071 ppm) (%) vs. exposition time (h)).
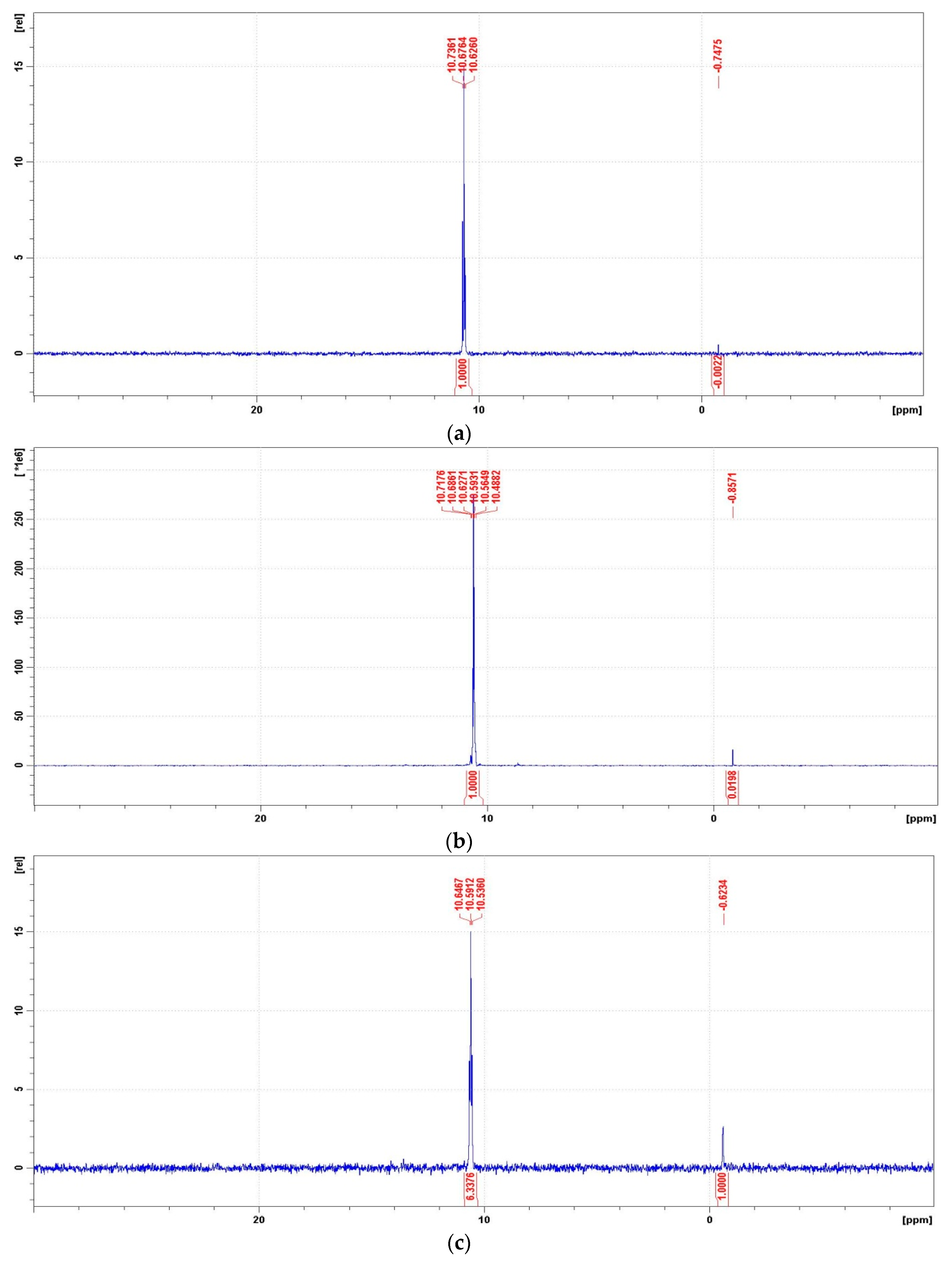
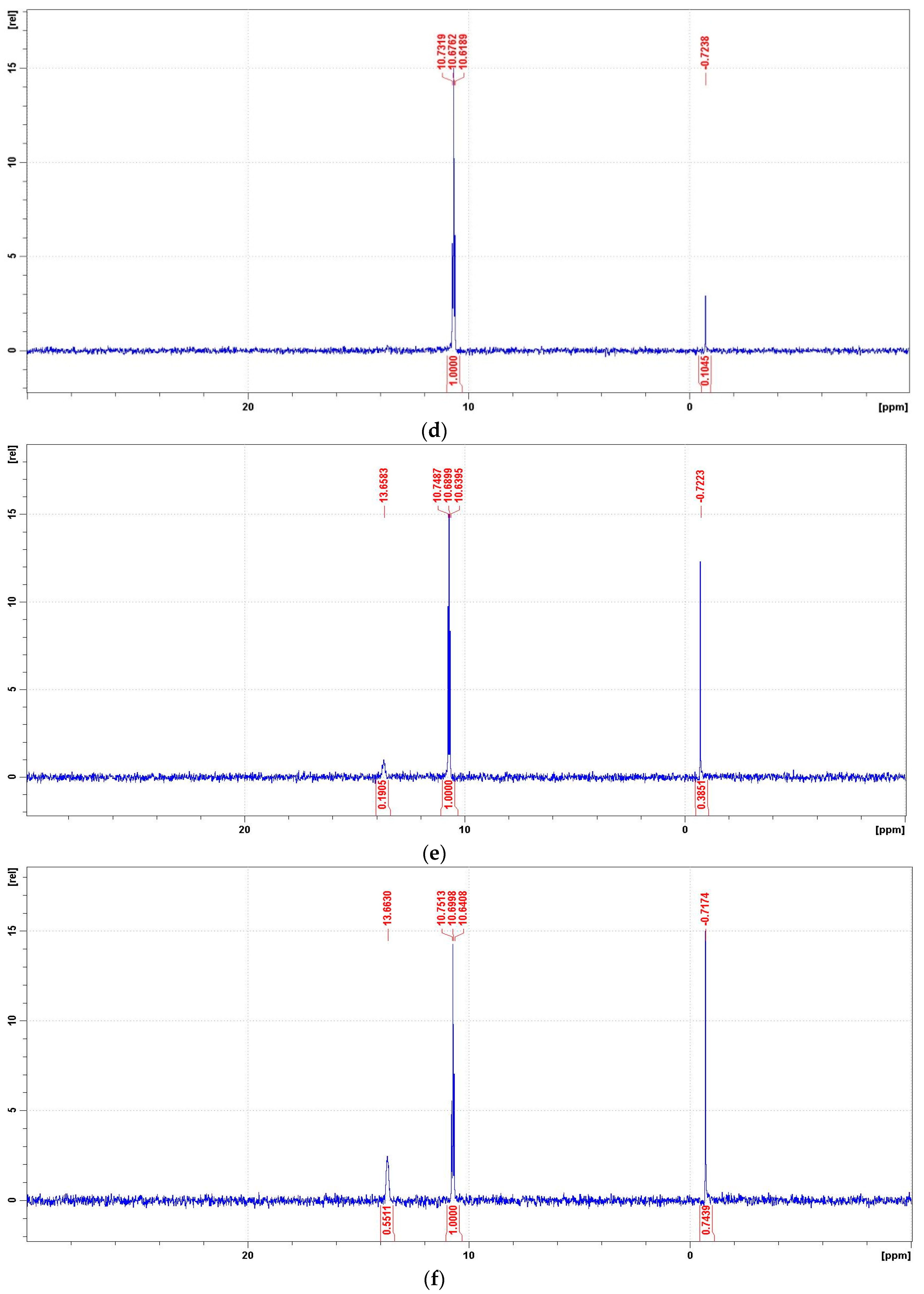
Figure 11.
Representative 31P NMR spectra of the degradation mixtures of PMG-H2O2-(UV) carried out at pH = 2–12 (UV; 25 °C), recorded after a given reaction time. (a) 31P NMR spectrum of the reaction mixture recorded before radiation (dissolved in 2 M H2SO4/D2SO4/); (b) reaction mixture after UV radiation at pH = 2 (reaction time 360 min); (c) reaction mixture after UV radiation at pH = 8 (reaction time 360 min); (d) reaction mixture after UV radiation at pH = 10 (reaction time 360 min); (e) reaction mixture after UV radiation at pH = 12 (reaction time 30 min); (f) reaction mixture after UV radiation at pH = 12 (reaction time 360 min).
Figure 11.
Representative 31P NMR spectra of the degradation mixtures of PMG-H2O2-(UV) carried out at pH = 2–12 (UV; 25 °C), recorded after a given reaction time. (a) 31P NMR spectrum of the reaction mixture recorded before radiation (dissolved in 2 M H2SO4/D2SO4/); (b) reaction mixture after UV radiation at pH = 2 (reaction time 360 min); (c) reaction mixture after UV radiation at pH = 8 (reaction time 360 min); (d) reaction mixture after UV radiation at pH = 10 (reaction time 360 min); (e) reaction mixture after UV radiation at pH = 12 (reaction time 30 min); (f) reaction mixture after UV radiation at pH = 12 (reaction time 360 min).
Figure 12.
Possible degradation paths of PMG (reaction time: 48 h).
Figure 12.
Possible degradation paths of PMG (reaction time: 48 h).
Table 1.
Representative applications of advanced oxidation process (AOP) technology in the chemical degradation of N-(PhosphonoMethyl)Glycine (PMG) and aminomethylphosphonic acid (GlyP).
Table 1.
Representative applications of advanced oxidation process (AOP) technology in the chemical degradation of N-(PhosphonoMethyl)Glycine (PMG) and aminomethylphosphonic acid (GlyP).
| No | P-Herbicide | AOP Systems | Phosphorus Degradation Products | Analysis | Reference |
|---|
| 1 | PMG and GlyP | Birnessite (Mn4+ and Mn3+) | H3PO4 | VIS (P-Mo Blue) | [20] |
| 2 | PMG | Fe(III)(C2O4)nm−/UV (365 nm) | H3PO4 | VIS (P-Mo Blue) | [21] |
| 3 | PMG | TiO2/UV (312 nm) | H3PO4 | HPLC-UV | [22] |
| 4 | PMG | O2/TiO2/SiO2/UV (365 nm) | H3PO4 | TOC, HPLC | [23] |
| 5 | PMG and GlyP | O3 (pH = 6.5 and 10) | H3PO4 | TOC, HPLC-FD | [24] |
| Photolysis (292 nm) | H3PO4 | TOC, HPLC-FD |
| TiO2/O2 (292 nm) | H3PO4 | TOC, HPLC-FD |
| 6 | PMG | H2O2/UV (254 nm) | H3PO4 | HPLC-FD | [25] |
| 7 | PMG | Birnessite (Mn4+ and Mn3+) | H3PO4 + GlyP + C-Px | 31P NMR | [26] |
Table 2.
Names, abbreviations, and structures of aminophosphonic acids discussed in this work a.
Table 2.
Names, abbreviations, and structures of aminophosphonic acids discussed in this work a.
| Structure | Name | Trivial Name | Abbr. | Synth. |
 | 1-aminoalkylphosphonic acid | phosphono amino acid | AAP |
| No | R | R1 |
| 1 | H | H | aminomethylphosphonic acid | phosphono-glycine | GlyP | [35] |
| | Me | H | (N-methylamino)methyl-phosphonic acid | phosphono-(N-methyl)-glycine | Me-GlyP | [37] |
| | Me | Me | (N,N-dimethylamino)-methylphosphonic acid | phosphono-(N,N-dimethyl)-glycine | Me2-GlyP | |
Table 3.
Preparation of the reaction mixtures for PMG-H2O2 and PMG-H2O2-Fe2+ systems.
Table 3.
Preparation of the reaction mixtures for PMG-H2O2 and PMG-H2O2-Fe2+ systems.
| Exp. | PMG
0.1 mmol | H2O | 10 M H2O2 | FeSO4 |
|---|
| 0.5 mmol | 1 mmol | 0.01 M | 0.02 M |
|---|
| 1 | 17 mg | 0.55 mL | 0.05 mL | − | − | − |
| 2 | 17 mg | 0.5 mL | − | 0.1 mL | − | − |
| 3 | 17 mg | − | − | 0.1 mL | 0.5 mL
(0.005 mmol) | − |
| 4 | 17 mg | − | − | 0.1 mL | − | 0.5 mL
(0.01 mmol) |
Table 4.
Preparation of the reaction mixtures for AOP degradations of PMG in Roundup herbicide.
Table 4.
Preparation of the reaction mixtures for AOP degradations of PMG in Roundup herbicide.
| No | Roundup
(0.75 M) | H2O | H2O2
(30%; 10 M) | H2SO4
(2 M) | NaOH (5 M) | pH |
|---|
| mL | PMG mmoL | mL | mL | mmoL | mL | mL |
|---|
| 1 | 16.0 | 12.0 | 3 680 | − | − | − | − | 4.85 |
| 2 | 16.0 | 12.0 | 3 680 | 6.0 | 60.0 | 4.6 | − | 2.0 |
| 3 | 16.0 | 12.0 | 3 680 | 6.0 | 60.0 | 0.5 | − | 4.0 |
| 4 | 16.0 | 12.0 | 3 680 | 6.0 | 60.0 | − | 4.0 | 6.0 |
| 5 | 16.0 | 12.0 | 3 680 | 6.0 | 60.0 | − | 4.8 | 8.0 |
| 6 | 16.0 | 12.0 | 3 680 | 6.0 | 60.0 | − | 6.0 | 10.0 |
| 7 | 16.0 | 12.0 | 3 680 | 6.0 | 60.0 | − | 40.0 | 12.0 |
Table 5.
Representative works on pKa determination of PMG.
Table 5.
Representative works on pKa determination of PMG.
| No. | pK | Method | Reference |
|---|
| pK1 | pK2 | pK3 | pK4 |
|---|
| 1 | 2.0 | 2.6 | 5.6 | 10.6 | pH metric titration | [40] |
| 2 | | 2.32 | 5.86 | 10.86 | pH metric titration | [41] |
| 3 | <1 | 2.0 | 5.5 | 10.5 | 1H and 31P NMR | [42] |
| 4 | 0.3 | 2.3 | 5.6 | 10.6 | 1H and 31P NMR | [43] |
| 5 | | 2.09 | 5.52 | 10.28 | pH metric titration | [44] |
| 6 | | logβ 9.66
(1.58) | logβ 14.86
(5.20) | log β 16.44
(9.66) | pH metric titration | [45] |
Table 6.
31P Chemical shifts (δ(ppm)) of PMG and its potential degradation products in acidic and basic solutions.
Table 6.
31P Chemical shifts (δ(ppm)) of PMG and its potential degradation products in acidic and basic solutions.
| 2 M HCl |
| Comp. | PMG | GlyP | Me-GlyP | Me2-GlyP | Me-PO3H2 | H3PO4 | H3PO3 |
| δ (31P) (ppm) | 10.6 | 13.9 | 11.4 | 9.4 | 30.7 | −0.47 | 5.15 |
| 2 M KOH |
| Comp. | PMG | GlyP | Me-GlyP | Me2-GlyP | Me-PO3H2 | H3PO4 | H3PO3 |
| δ (31P) (ppm) | 16.3 | 19.3 | 16.0 | 15.0 | 20.5 | 5.4 | 3.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
