Spatial Pattern of Bacterial Community Diversity Formed in Different Groundwater Field Corresponding to Electron Donors and Acceptors Distributions at a Petroleum-Contaminated Site


Abstract
1. Introduction
2. Materials and Methods
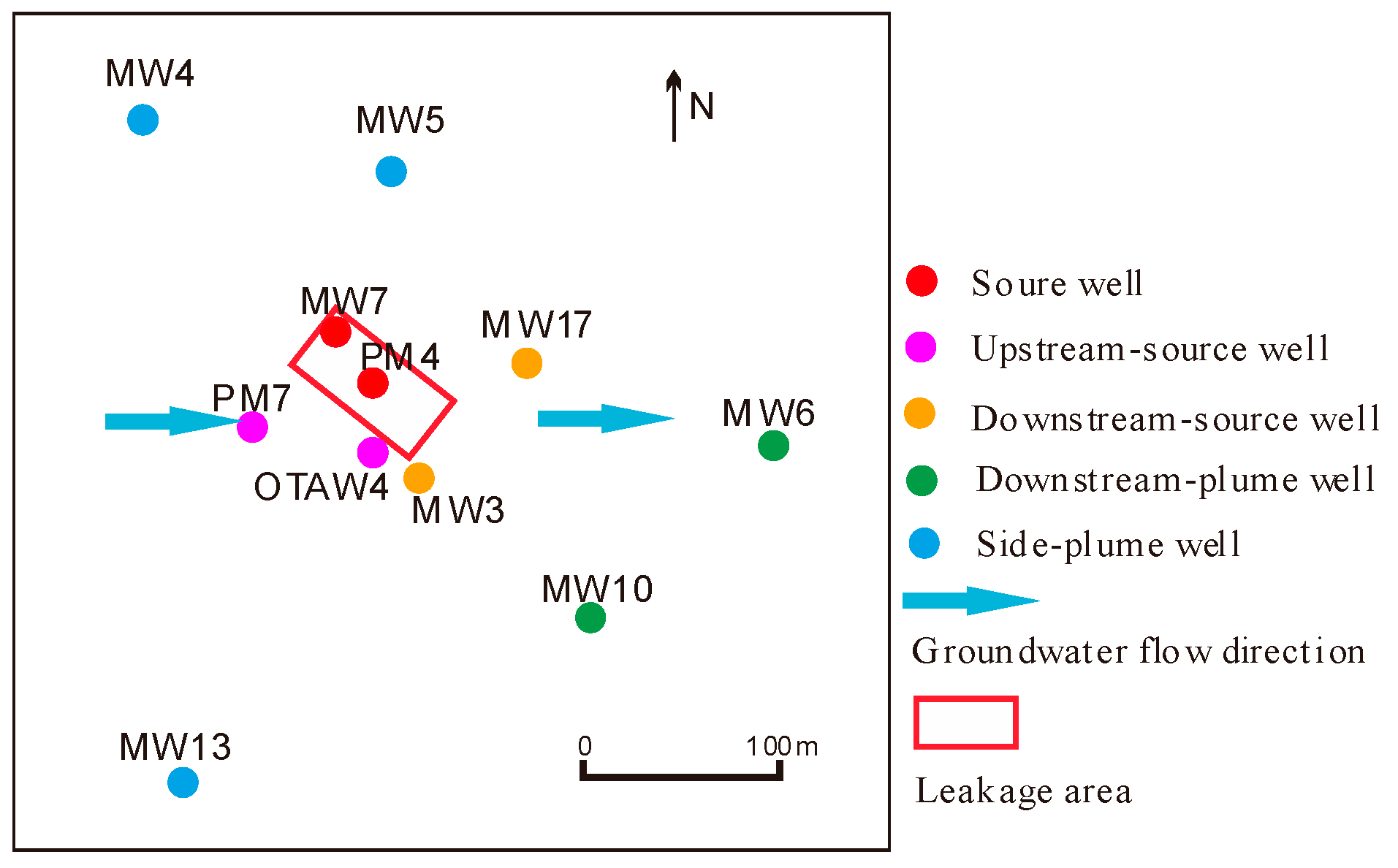
2.1. Site Description and Sampling Procedure
2.2. Chemical Analyses
2.3. DNA Extraction, PCR Amplification, Library Construction and Sequencing
2.4. Bioinformatics Analysis
3. Results
3.1. The Distribution of Electron Acceptors-Donors and Other Chemical Parameters
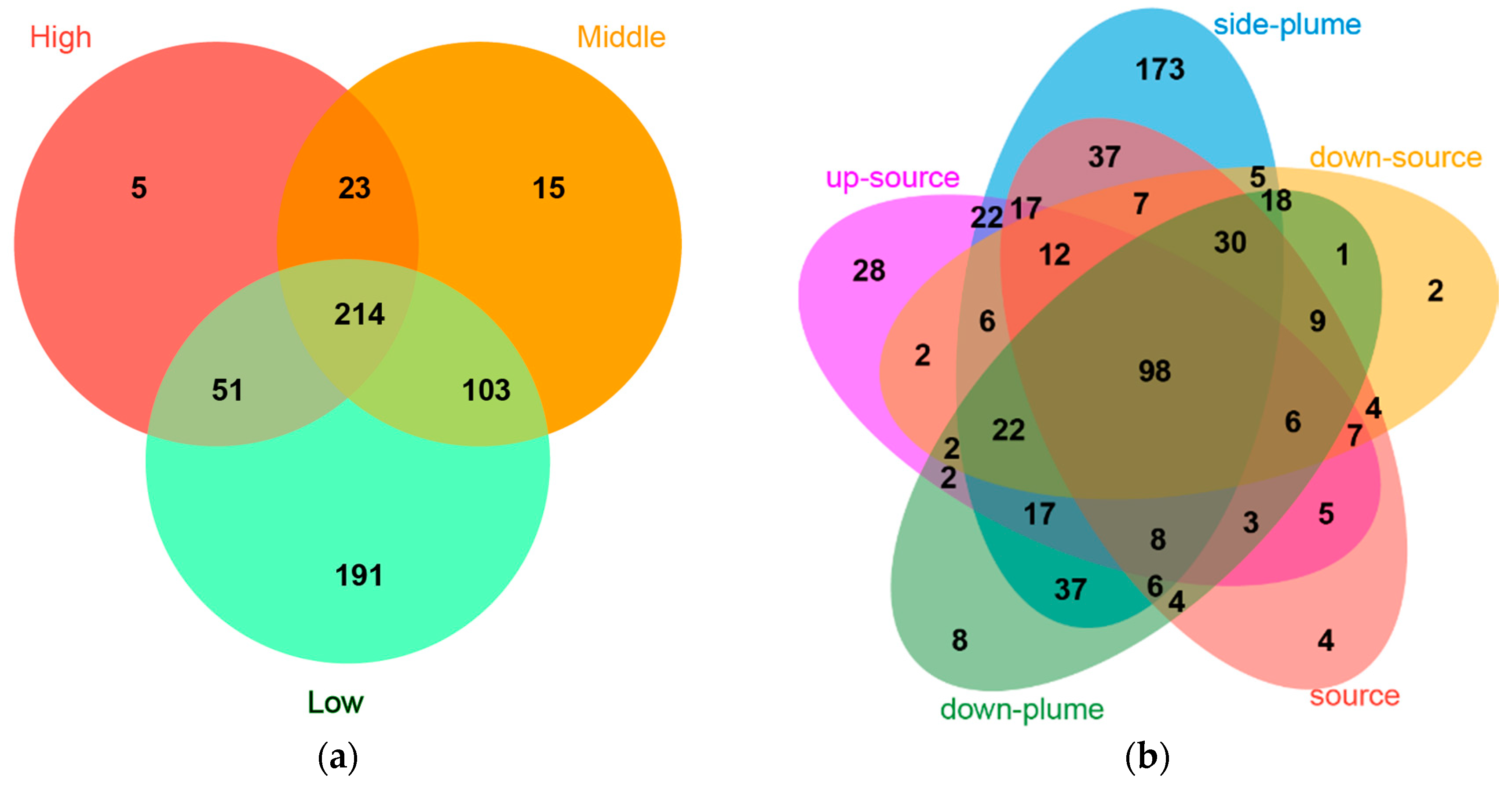
3.2. Alpha-Diversity Indexes
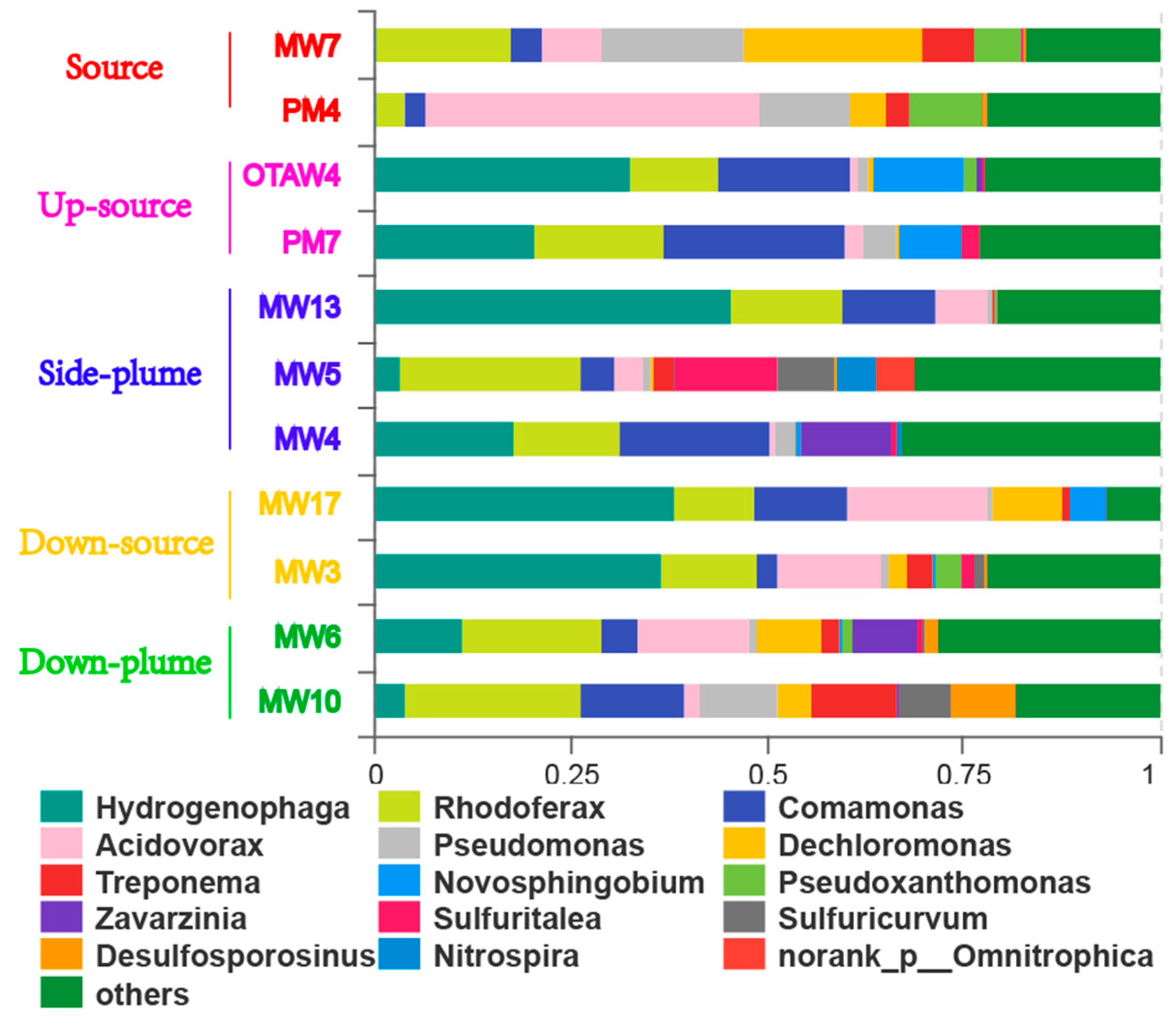
3.3. Community Composition
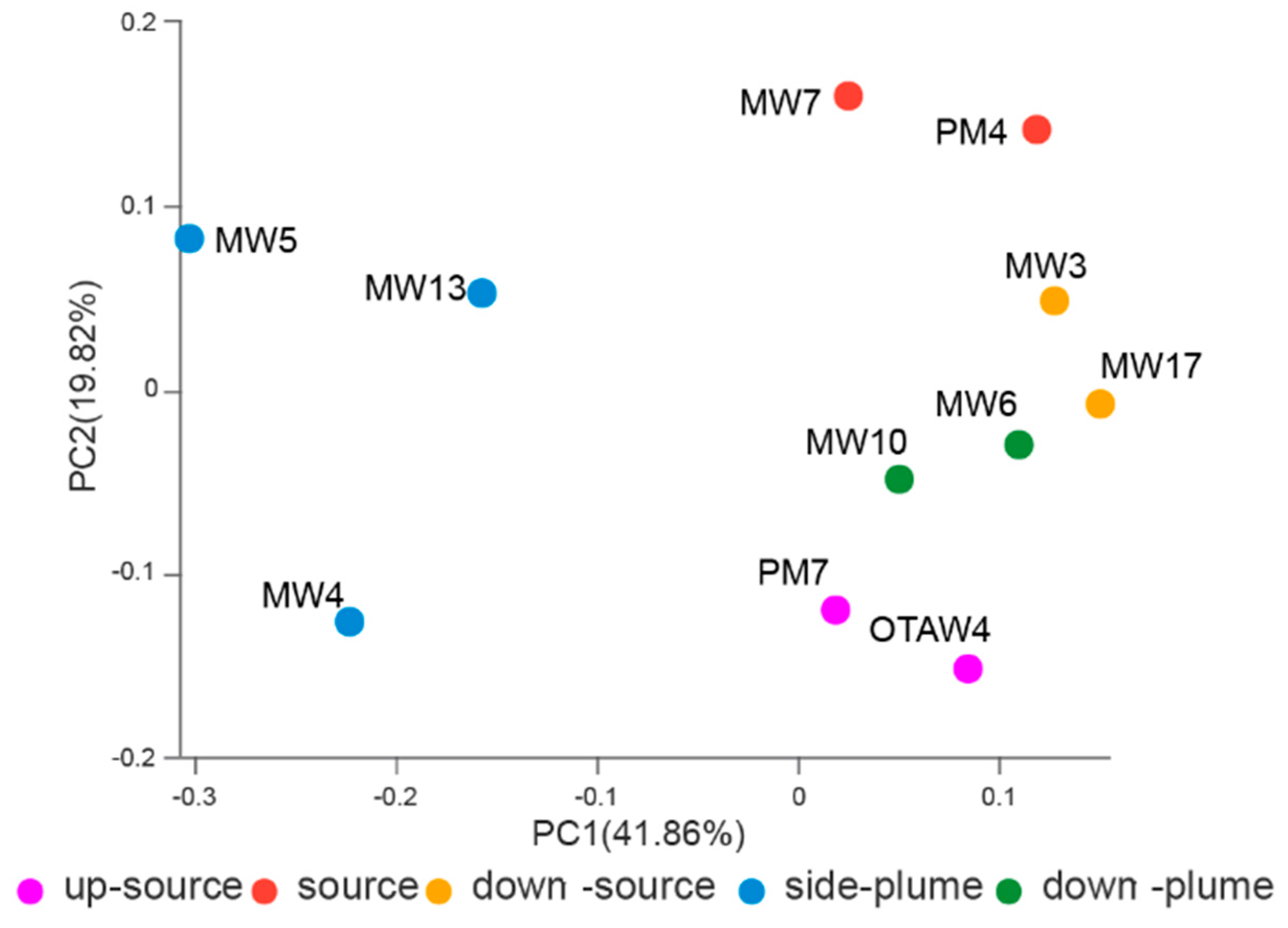
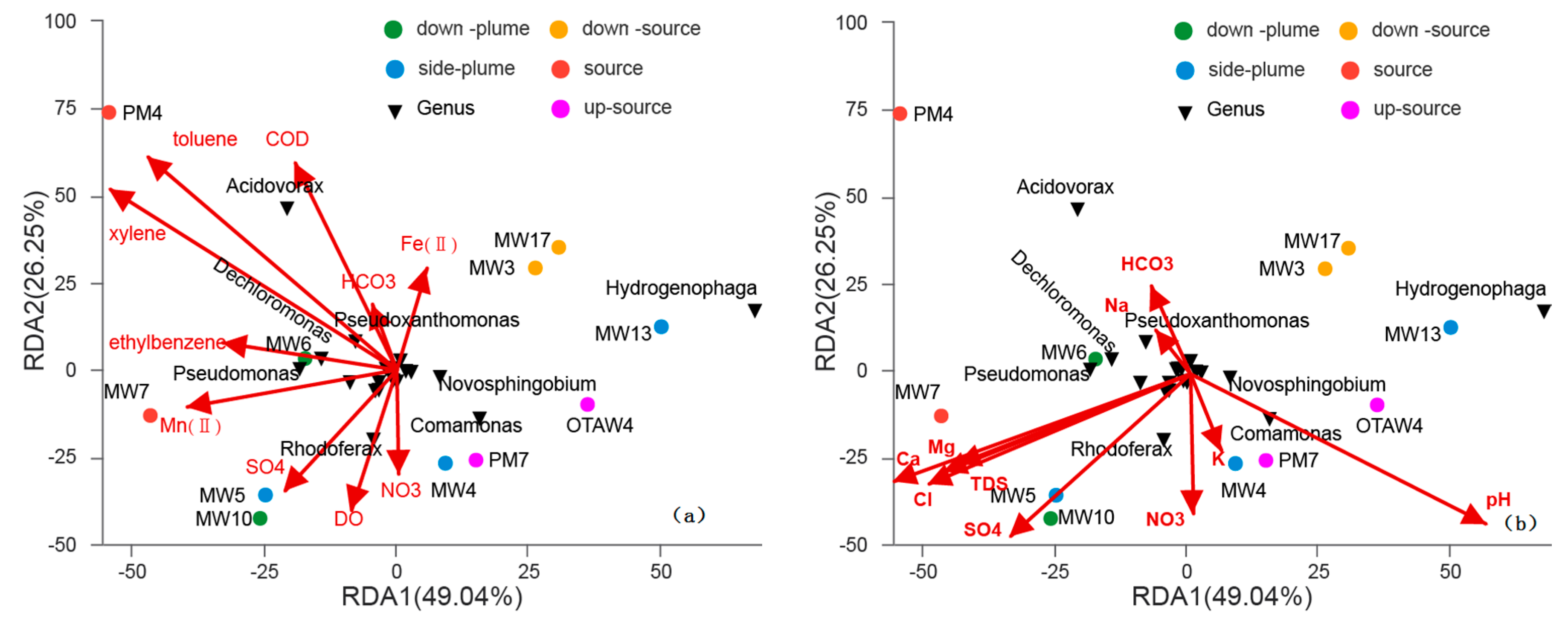
3.4. Relationships between Bacterial Communities among Samples
4. Discussion
4.1. Variations in Bacterial Communities with Electron Donor Concentrations
4.2. The Influence of Electron Acceptors on Bacterial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shahi, A.; Ince, B.; Aydin, S.; Ince, O. Assessment of the horizontal transfer of functional genes as a suitable approach for evaluation of the bioremediation potential of petroleum-contaminated sites: A mini-review. Appl. Microbiol. Biotechnol. 2017, 101, 4341–4348. [Google Scholar] [CrossRef] [PubMed]
- López, E.; Schuhmacher, M.; Domingo, J.L. Human health risks of petroleum-contaminated groundwater. Environ. Sci. Pollut. Res. 2008, 15, 278–288. [Google Scholar] [CrossRef]
- Zhang, S.; Su, X.; Lin, X.; Zhang, Y.; Zhang, Y. Experimental study on the multi-media prb reactor for the remediation of petroleum-contaminated groundwater. Environ. Earth Sci. 2015, 73, 5611–5618. [Google Scholar] [CrossRef]
- Hunkeler, D.; Höhener, P.; Bernasconi, S.; Zeyer, J. Engineered in situ bioremediation of a petroleum hydrocarbon-contaminated aquifer: Assessment of mineralization based on alkalinity, inorganic carbon and stable carbon isotope balances. J. Contam. Hydrol. 1999, 37, 201–223. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2009, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Schiegg, H.-O. Field infiltration as a method for the disposal of oil-in-water emulsions from the restoration of oil-polluted aquifers. Water Res. 1980, 14, 1011–1016. [Google Scholar] [CrossRef]
- Tischer, K.; Kleinsteuber, S.; Schleinitz, K.M.; Fetzer, I.; Spott, O.; Stange, F.; Lohse, U.; Franz, J.; Neumann, F.; Gerling, S.; et al. Microbial communities along biogeochemical gradients in a hydrocarbon-contaminated aquifer. Environ. Microbiol. 2013, 15, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.J.; Aislabie, J.M.; Brown, C.E.; Harris, L.; Foght, J.M. Hydrocarbon contamination changes the bacterial diversity of soil from around scott base, antarctica. FEMS Microbiol. Ecol. 2005, 53, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Abed, R.M.M.; Al-Kindi, S.; Al-Kharusi, S. Diversity of bacterial communities along a petroleum contamination gradient in desert soils. Microb. Ecol. 2015, 69, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Zi, X.; Wang, Q. Bacterial community diversity of oil-contaminated soils assessed by high throughput sequencing of 16s rRNA genes. Int. J. Environ. Res. Public Health 2015, 12, 12002–12015. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Huggins, T.; Jin, S.; Zuo, Y.; Ren, Z.J. Microbial metabolism and community structure in response to bioelectrochemically enhanced remediation of petroleum hydrocarbon-contaminated soil. Environ. Sci. Technol. 2014, 48, 4021–4029. [Google Scholar] [CrossRef] [PubMed]
- Fahy, A.; Lethbridge, G.; Earle, R.; Ball, A.S.; Timmis, K.N.; McGenity, T.J. Effects of long-term benzene pollution on bacterial diversity and community structure in groundwater. Environ. Microbiol. 2005, 7, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, Y.; Su, X.; Cui, X. Responses of hydrochemical parameters, community structures, and microbial activities to the natural biodegradation of petroleum hydrocarbons in a groundwater–soil environment. Environ. Earth Sci. 2016, 75, 1400. [Google Scholar] [CrossRef]
- Zhou, A.-X.; Zhang, Y.-L.; Dong, T.-Z.; Lin, X.-Y.; Su, X.-S. Response of the microbial community to seasonal groundwater level fluctuations in petroleum hydrocarbon-contaminated groundwater. Environ. Sci. Pollut. Res. 2015, 22, 10094–10106. [Google Scholar] [CrossRef] [PubMed]
- Lueders, T. The ecology of anaerobic degraders of btex hydrocarbons in aquifers. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Yergeau, E.; Sanschagrin, S.; Maynard, C.; St-Arnaud, M.; Greer, C.W. Microbial expression profiles in the rhizosphere of willows depend on soil contamination. ISME J. 2013, 8, 344. [Google Scholar] [CrossRef] [PubMed]
- Main, C.E.; Ruhl, H.A.; Jones, D.O.B.; Yool, A.; Thornton, B.; Mayor, D.J. Hydrocarbon contamination affects deep-sea benthic oxygen uptake and microbial community composition. Deep Sea Res. Part I Oceanogr. Res. Pap. 2015, 100, 79–87. [Google Scholar] [CrossRef]
- Dorer, C.; Vogt, C.; Neu, T.R.; Stryganyuk, G.; Richnow, H. Characterization of toluene and ethylbenzene biodegradation under nitrate-, iron(III)- and manganese(IV)-reducing conditions by compound-specific isotope analysis. Environ. Pollut. 2016, 211, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Acosta-González, A.; Marqués, S. Bacterial diversity in oil-polluted marine coastal sediments. Curr. Opin. Biotechnol. 2016, 38, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Gieg, L.M.; Fowler, S.J.; Berdugo-Clavijo, C. Syntrophic biodegradation of hydrocarbon contaminants. Curr. Opin. Biotechnol. 2014, 27, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Meckenstock, R.U.; Elsner, M.; Griebler, C.; Lueders, T.; Stumpp, C.; Aamand, J.; Agathos, S.N.; Albrechtsen, H.-J.; Bastiaens, L.; Bjerg, P.L.; et al. Biodegradation: Updating the concepts of control for microbial cleanup in contaminated aquifers. Environ. Sci. Technol. 2015, 49, 7073–7081. [Google Scholar] [CrossRef] [PubMed]
- Röling, W.F.M.; van Breukelen, B.M.; Braster, M.; Lin, B.; van Verseveld, H.W. Relationships between microbial community structure and hydrochemistry in a landfill leachate-polluted aquifer. Appl. Environ. Microbiol. 2001, 67, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Gu, J.; Zhao, Q.; Liu, Y. Cod capture: A feasible option towards energy self-sufficient domestic wastewater treatment. Sci. Rep. 2016, 6, 25054. [Google Scholar] [CrossRef] [PubMed]
- United States Environmental Protection Agency (U.S.E.P). Method 8260b Volatile Organic Compounds by Gas Chromatography/Mass Spectrometry (gc/ms); United States Environmental Protection Agency: Washington, DC, USA, 1996.
- Standard, A. Methods for the Examination of Water and Wastewate; American Public Health Association: Washington, DC, USA, 1998. [Google Scholar]
- Ye, J.; Song, Z.; Wang, L.; Zhu, J. Metagenomic analysis of microbiota structure evolution in phytoremediation of a swine lagoon wastewater. Bioresour. Technol. 2016, 219, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Li, F.; Wang, X.; Zhang, X.; Liu, T.; Nian, F.; Yue, X.; Li, F.; Pan, X.; et al. Effects of early feeding on the host rumen transcriptome and bacterial diversity in lambs. Sci. Rep. 2016, 6, 32479. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yao, J.; Ainiwaer, M.; Hong, Y.; Zhang, Y. Analysis of bacterial community structure of activated sludge from wastewater treatment plants in winter. BioMed Res. Int. 2018, 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Qiu, S.; Zhang, J.; Xia, C. Study on hydrogenophaga palleronii LHJ38—A naphthalene-degrading strain with high activity. Environ. Prot. Chem. Ind. 2006, 26, 87–90. [Google Scholar]
- Yang, Q.; Cai, S.; Dong, S.; Chen, L.; Chen, J.; Cai, T. Biodegradation of 3-methyldiphenylether (MDE) by hydrogenophaga atypical strain QY7-2 and cloning of the methy-oxidation gene mdeabcd. Sci. Rep. 2016, 6, 39270. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.K.; Zylstra, G.J. Molecular cloning of novel genes for polycyclic aromatic hydrocarbon degradation from Comamonas testosteroni GZ39. Appl. Environ. Microbiol. 1996, 62, 230–236. [Google Scholar] [PubMed]
- Aburto, A.; Peimbert, M. Degradation of a benzene–toluene mixture by hydrocarbon-adapted bacterial communities. Ann. Microbiol. 2011, 61, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.R.; Guzmán Ramirez, L.; Aitken, M.D. Characterization of a polycyclic aromatic hydrocarbon degradation gene cluster in a phenanthrene-degrading acidovorax strain. Appl. Environ. Microbiol. 2009, 75, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.M.; Karl, M.R.; Blatt, L.M.; Simon, M.J.; Winter, R.B.; Fausset, P.R.; Lu, H.S.; Harcourt, A.A.; Chen, K.K. Cloning and characterization of a pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 1991, 173, 5315–5327. [Google Scholar] [CrossRef] [PubMed]
- Liebensteiner, M.G.; Oosterkamp, M.J.; Stams, A.J.M. Microbial respiration with chlorine oxyanions: Diversity and physiological and biochemical properties of chlorate- and perchlorate-reducing microorganisms. Ann. N. Y. Acad. Sci. 2016, 1365, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Mehboob, F.; Weelink, S.; Saia, F.T.; Junca, H.; Stams, A.J.M.; Schraa, G. Microbial degradation of aliphatic and aromatic hydrocarbons with (per)chlorate as electron acceptor. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 935–945. [Google Scholar]
- Nayak, A.S.; Vijaykumar, M.H.; Karegoudar, T.B. Characterization of biosurfactant produced by Pseudoxanthomonas sp. PNK-04 and its application in bioremediation. Int. Biodeterior. Biodegrad. 2009, 63, 73–79. [Google Scholar] [CrossRef]
- Kim, J.M.; Le, N.T.; Chung, B.S.; Park, J.H.; Bae, J.-W.; Madsen, E.L.; Jeon, C.O. Influence of soil components on the biodegradation of benzene, toluene, ethylbenzene, and o-, m-, and p-xylenes by the newly isolated bacterium pseudoxanthomonas spadix BD-a59. Appl. Environ. Microbiol. 2008, 74, 7313–7320. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.H.; Kwon, K.K.; Kang, J.-H.; Jung, H.-B.; Kim, S.-J. Novosphingobium pentaromativorans sp. Nov., a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment. Int. J. Syst. Evol. Microbiol. 2004, 54, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-P.; Wang, B.-J.; Liu, Y.-H.; Liu, S.-J. Novosphingobium taihuense sp. Nov., a novel aromatic-compound-degrading bacterium isolated from taihu lake, china. Int. J. Syst. Evol. Microbiol. 2005, 55, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, A.V.; Davidova, I.A.; Savage-Ashlock, K.; Parisi, V.A.; Gieg, L.M.; Suflita, J.M.; Kukor, J.J.; Wawrik, B. Diversity of benzyl- and alkylsuccinate synthase genes in hydrocarbon-impacted environments and enrichment cultures. Environ. Sci. Technol. 2010, 44, 7287–7294. [Google Scholar] [CrossRef] [PubMed]
- Rochman, F.F.; Sheremet, A.; Tamas, I.; Saidi-Mehrabad, A.; Kim, J.-J.; Dong, X.; Sensen, C.W.; Gieg, L.M.; Dunfield, P.F. Benzene and naphthalene degrading bacterial communities in an oil sands tailings pond. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, X.; Song, T.; Yu, Y.; Qi, J. Stimulation effect of electric current density (ECD) on microbial community of a three dimensional particle electrode coupled with biological aerated filter reactor (TDE-BAF). Bioresour. Technol. 2017, 243, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.U.; Zhang, F.; Zhong-Qi, X.U.; Jia, Y.L.; You, J.M. Some characteristics of simpson index and the shannon-wiener index and their dilution effect. Pratacult. Sci. 2011, 28, 527–531. [Google Scholar]
- Sutton, N.B.; Maphosa, F.; Morillo, J.A.; Abu Al-Soud, W.; Langenhoff, A.A.M.; Grotenhuis, T.; Rijnaarts, H.H.M.; Smidt, H. Impact of long-term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 2013, 79, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, R.; Yu, Z.; Zhang, H.; Yun, J.; Li, Y.; Liu, X.; Pan, J. Pyrosequencing reveals the dominance of methylotrophic methanogenesis in a coal bed methane reservoir associated with eastern ordos basin in china. Int. J. Coal Geol. 2012, 93, 56–61. [Google Scholar] [CrossRef]
- Jones, E.J.P.; Voytek, M.A.; Corum, M.D.; Orem, W.H. Stimulation of methane generation from nonproductive coal by addition of nutrients or a microbial consortium. Appl. Environ. Microbiol. 2010, 76, 7013–7022. [Google Scholar] [CrossRef] [PubMed]
- Van Eerten-Jansen, M.C.A.A.; Veldhoen, A.B.; Plugge, C.M.; Stams, A.J.M.; Buisman, C.J.N.; Ter Heijne, A. Microbial community analysis of a methane-producing biocathode in a bioelectrochemical system. Archaea 2013, 2013, 481784. [Google Scholar] [CrossRef] [PubMed]
- Hersman, L.E.; Huang, A.; Maurice, P.A.; Forsythe, J.H. Siderophore production and iron reduction by pseudomonas mendocina in response to iron deprivation. Geomicrobiol. J. 2000, 17, 261–273. [Google Scholar]
- Pantke, C.; Obst, M.; Benzerara, K.; Morin, G.; Ona-Nguema, G.; Dippon, U.; Kappler, A. Green rust formation during fe(ii) oxidation by the nitrate-reducing Acidovorax sp. Strain bofen1. Environ. Sci. Technol. 2012, 46, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Coates, J.D. Hydroxylation and carboxylation—Two crucial steps of anaerobic benzene degradation by dechloromonas strain rcb. Appl. Environ. Microbiol. 2005, 71, 5427–5432. [Google Scholar] [CrossRef] [PubMed]
- Finneran, K.T.; Johnsen, C.V.; Lovley, D.R. Rhodoferax ferrireducens sp. Nov., a psychrotolerant, facultatively anaerobic bacterium that oxidizes acetate with the reduction of fe(iii). Int. J. Syst. Evol. Microbiol. 2003, 53, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Patricia, A.M.; Vierkorn, M.A.; Hersman, L.E.; Fulghum, J.E.; Ferryman, A. Enhancement of kaolinite dissolution by an aerobic pseudomonas mendocina bacterium. Geomicrobiol. J. 2001, 18, 21–35. [Google Scholar] [CrossRef]
- Jovanović, T.; Ascenso, C.; Hazlett, K.R.O.; Sikkink, R.; Krebs, C.; Litwiller, R.; Benson, L.M.; Moura, I.; Moura, J.J.G.; Radolf, J.D.; et al. Neelaredoxin, an iron-binding protein from the syphilis spirochete, treponema pallidum, is a superoxide reductase. J. Biol. Chem. 2000, 275, 28439–28448. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Sass, H.; Langner, H.; Schumann, P.; Kroppenstedt, R.M.; Spring, S.; Overmann, J.; Rosenzweig, R.F. Desulfosporosinus lacus sp. Nov., a sulfate-reducing bacterium isolated from pristine freshwater lake sediments. Int. J. Syst. Evol. Microbiol. 2006, 56, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Fukui, M. Sulfuritalea hydrogenivorans gen. Nov., sp. Nov., a facultative autotroph isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 2011, 61, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Watanabe, K. Sulfuricurvum kujiense gen. Nov., sp. Nov., a facultatively anaerobic, chemolithoautotrophic, sulfur-oxidizing bacterium isolated from an underground crude-oil storage cavity. Int. J. Syst. Evol. Microbiol. 2004, 54, 2297–2300. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Lücker, S.; Albertsen, M.; Kitzinger, K.; Herbold, C.; Spieck, E.; Nielsen, P.H.; Wagner, M.; Daims, H. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus nitrospira. Proc. Natl. Acad. Sci. USA 2015, 112, 11371–11376. [Google Scholar] [CrossRef] [PubMed]
- Ludington, W.B.; Seher, T.D.; Applegate, O.; Li, X.; Kliegman, J.I.; Langelier, C.; Atwill, E.R.; Harter, T.; Derisi, J.L. Assessing biosynthetic potential of agricultural groundwater through metagenomic sequencing: A diverse anammox community dominates nitrate-rich groundwater. PLoS ONE 2017, 12, e0174930. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Zhang, S.D.; Zhang, W.J.; Rouy, Z.; Alberto, F.; Santini, C.L.; Mangenot, S.; Gagnot, S.; Philippe, N.; Pradel, N. The chimeric nature of the genomes of marine magnetotactic coccoid-ovoid bacteria defines a novel group of proteobacteria. Environ. Microbiol. 2017, 19, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Zhuang, L.; Zhou, S.-G.; Li, F.-B.; Li, X.-M. Fe(III)-enhanced anaerobic transformation of 2,4-dichlorophenoxyacetic acid by an iron-reducing bacterium Comamonas koreensis Cy01. FEMS Microbiol. Ecol. 2009, 71, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Patureau, D.; Bernet, N.; Moletta, R. Study of the denitrifying enzymatic system of Comamonas sp. Strain SGLY2 under various aeration conditions with a particular view on nitrate and nitrite reductases. Curr. Microbiol. 1996, 32, 25–32. [Google Scholar] [CrossRef]
- Kai, X.X.; Fang, M.C. A experimental study of influence of ph on calcium carbonate crystallization fouling. Petro-Chem. Equip. 2004, 33, 11–14. [Google Scholar]
- Marcus, D.N.; Pinto, A.; Anantharaman, K.; Ruberg, S.A.; Kramer, E.L.; Raskin, L.; Dick, G.J. Diverse manganese(II)-oxidizing bacteria are prevalent in drinking water systems. Environ. Microbiol. Rep. 2017, 9, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Mechichi, T.; Stackebrandt, E.; Fuchs, G. Alicycliphilus denitrificans gen. Nov., sp. Nov., a cyclohexanol-degrading, nitrate-reducing β-proteobacterium. Int. J. Syst. Evol. Microbiol. 2003, 53, 147–152. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Side-Plume | Upstream-Source | Source | Downstream-Source | Downstream-Plume | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Well | MW5 | MW4 | MW13 | PM7 | OTAW4 | MW7 | PM4 | MW3 | MW17 | MW6 | MW10 | |
| Electron donors (μg·L−1) | toluene | 97 | 4 | 6 | 315 | 108 | 11,211 | 20,610 | 723 | 11,680 | 7689 | 287 |
| ethylbenzene | 11.1 | 6.9 | 0.1 | 51.7 | 20 | 4316 | 583 | 832.9 | 2078 | 668 | 971.5 | |
| m(p)-xylene | 22.21 | 4.81 | 1.24 | 58.3 | 25 | 3001 | 3678 | 579.17 | 1636.5 | 1115 | 759.8 | |
| o-xylene | 22.22 | 2.13 | 0.66 | 56.67 | 8.3 | 3001 | 3680 | 320.2 | 1636.5 | 225 | 89.45 | |
| Electron acceptors (mg·L−1) | DO | 2.38 | 2.19 | 1.95 | 0.87 | 0.76 | 1.29 | 0.84 | 1.3 | 0.85 | 1.97 | 1.67 |
| NO3− | 16.77 | 68.96 | 4.6 | 1.78 | <0.20 | 1.75 | 1.76 | 1.76 | 1.75 | 11.5 | 7.73 | |
| SO42− | 107.2 | 277.6 | 39.67 | 38.15 | 69.22 | 83.79 | 66.7 | 21.54 | 16.68 | 163.3 | 98.7 | |
| HCO3− | 791.6 | 780.1 | 889.1 | 316.7 | 494.5 | 648.5 | 715.5 | 712.5 | 831.8 | 822.1 | 767.3 | |
| metabolic by-products (mg·L−1) | Fe2+ | 0.049 | 0.018 | 0.372 | 0.127 | 0.061 | 0.489 | 0.806 | 0.587 | 4.643 | 2.305 | 1.586 |
| Mn2+ | 6.48 | 0.625 | 1.953 | 0.755 | 0.781 | 2.356 | 2.5 | 1.473 | 2.589 | 3.518 | 2.862 | |
| Other ion (mg·L−1) | K+ | 3.14 | 3.35 | 2.49 | 1.58 | 1.91 | 2.2 | 1.72 | 1.43 | 2.17 | 2.57 | 1.01 |
| Na+ | 179.3 | 135.9 | 149.4 | 67.96 | 135.8 | 133.6 | 137.8 | 145.6 | 165.4 | 124.1 | 152.4 | |
| Ca2+ | 282.9 | 255.9 | 141.2 | 61.56 | 99.63 | 151.4 | 162.3 | 108.9 | 139.4 | 209.4 | 172.8 | |
| Mg2+ | 121.4 | 93.74 | 61.81 | 21.59 | 42.24 | 62.44 | 59.96 | 41.58 | 61.32 | 81.87 | 60.19 | |
| Cl− | 583.6 | 214.6 | 134.5 | 61.32 | 162.7 | 198.6 | 195.1 | 141.8 | 191.6 | 182.9 | 193.4 | |
| TDS | 1675 | 1439 | 975.3 | 412.3 | 758.8 | 958.1 | 983.1 | 818.9 | 994.3 | 1177 | 1064 | |
| COD (mg·L−1) | 4.37 | 1.65 | 4.54 | 6.19 | 56.23 | 162.4 | 268.7 | 56.23 | 337.4 | 40.63 | 31.24 | |
| pH | 6.94 | 7.26 | 7.22 | 7.59 | 7.66 | 7.15 | 7.02 | 7.22 | 6.94 | 7.15 | 7.28 | |
| Location | Sample | Diversity Indexes | |||||
|---|---|---|---|---|---|---|---|
| Sobs | Shannon | Simpson | Ace | Chao | Coverage | ||
| Side-plume | MW5 | 383 | 3.73 | 0.07 | 428.3 | 443.86 | 0.995 |
| MW4 | 324 | 3.59 | 0.08 | 356.17 | 358.89 | 0.996 | |
| MW13 | 249 | 2.82 | 0.12 | 317.9 | 314.27 | 0.995 | |
| Upstream-source | PM7 | 211 | 2.89 | 0.12 | 260.23 | 262.21 | 0.996 |
| OTAW4 | 191 | 2.76 | 0.15 | 240.13 | 226.25 | 0.997 | |
| Source | MW7 | 197 | 2.79 | 0.12 | 246.43 | 238.44 | 0.996 |
| PM4 | 186 | 2.7 | 0.2 | 341.12 | 294.48 | 0.995 | |
| Downstream-source | MW3 | 190 | 2.61 | 0.17 | 287.61 | 263.75 | 0.995 |
| MW17 | 160 | 2.18 | 0.2 | 204.92 | 196.96 | 0.997 | |
| Downstream-plume | MW6 | 215 | 3.44 | 0.07 | 266.41 | 291.56 | 0.996 |
| MW10 | 204 | 3.05 | 0.09 | 256.63 | 255.11 | 0.996 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, Z.; Zhang, M.; He, Z.; Cai, P.; Guo, C.; Wang, P. Spatial Pattern of Bacterial Community Diversity Formed in Different Groundwater Field Corresponding to Electron Donors and Acceptors Distributions at a Petroleum-Contaminated Site. Water 2018, 10, 842. https://doi.org/10.3390/w10070842
Ning Z, Zhang M, He Z, Cai P, Guo C, Wang P. Spatial Pattern of Bacterial Community Diversity Formed in Different Groundwater Field Corresponding to Electron Donors and Acceptors Distributions at a Petroleum-Contaminated Site. Water. 2018; 10(7):842. https://doi.org/10.3390/w10070842
Chicago/Turabian StyleNing, Zhuo, Min Zhang, Ze He, Pingping Cai, Caijuan Guo, and Ping Wang. 2018. "Spatial Pattern of Bacterial Community Diversity Formed in Different Groundwater Field Corresponding to Electron Donors and Acceptors Distributions at a Petroleum-Contaminated Site" Water 10, no. 7: 842. https://doi.org/10.3390/w10070842
APA StyleNing, Z., Zhang, M., He, Z., Cai, P., Guo, C., & Wang, P. (2018). Spatial Pattern of Bacterial Community Diversity Formed in Different Groundwater Field Corresponding to Electron Donors and Acceptors Distributions at a Petroleum-Contaminated Site. Water, 10(7), 842. https://doi.org/10.3390/w10070842