The Impact of Divalent Cations on the Enrichment of Soluble Saccharides in Primary Sea Spray Aerosol


Abstract
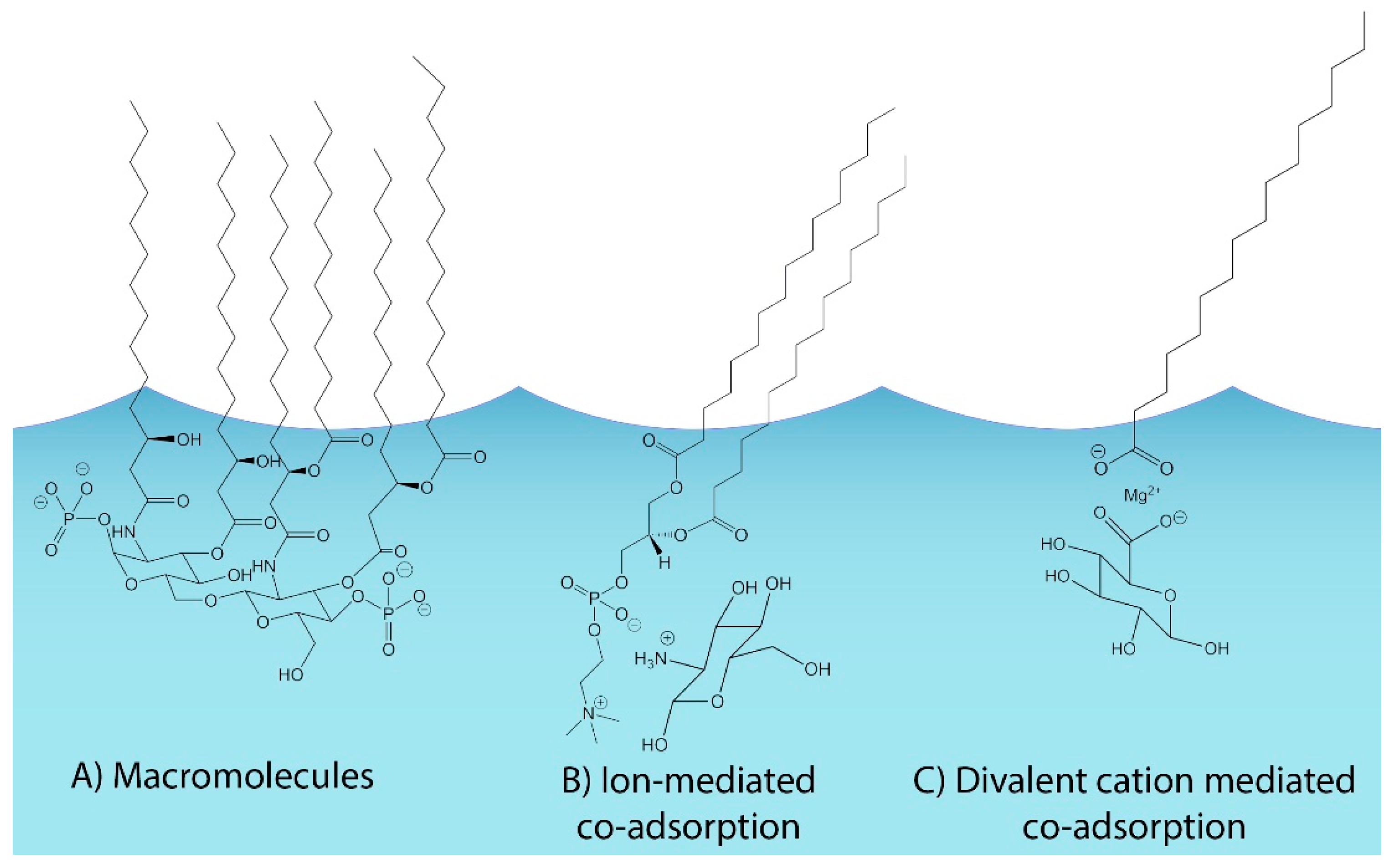
1. Introduction
2. Experiments
2.1. Generation of Synthetic Sea Spray Aerosol
2.2. Determination of Aerosol Supersaturated Hygroscopicity
2.3. Ion Exchange Chromatography Analysis
3. Results
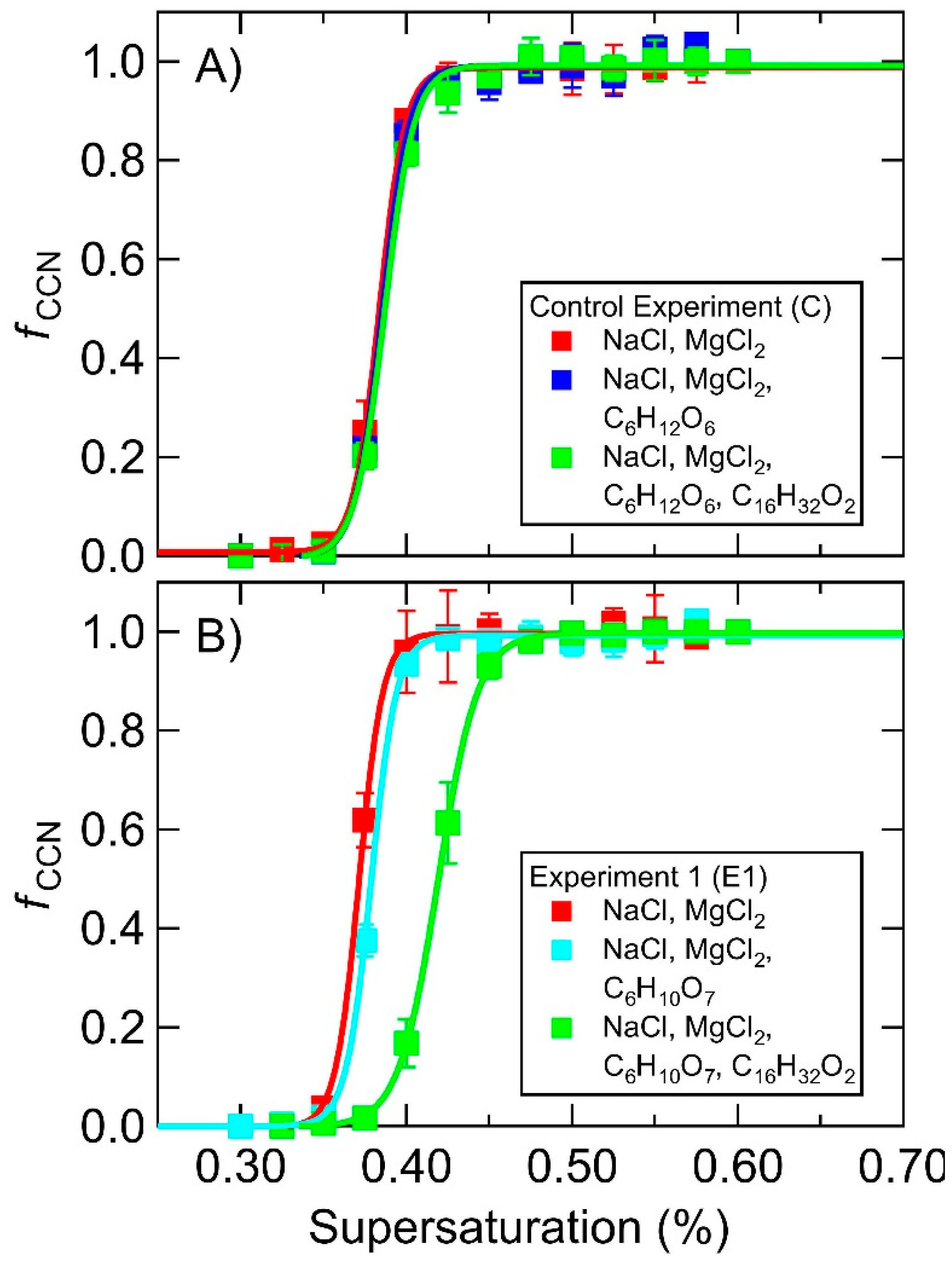
3.1. Supersaturated Hygroscopicity of Pure SSA Mimics
3.2. Supersaturated Hygroscopicity of MART Generated SSA
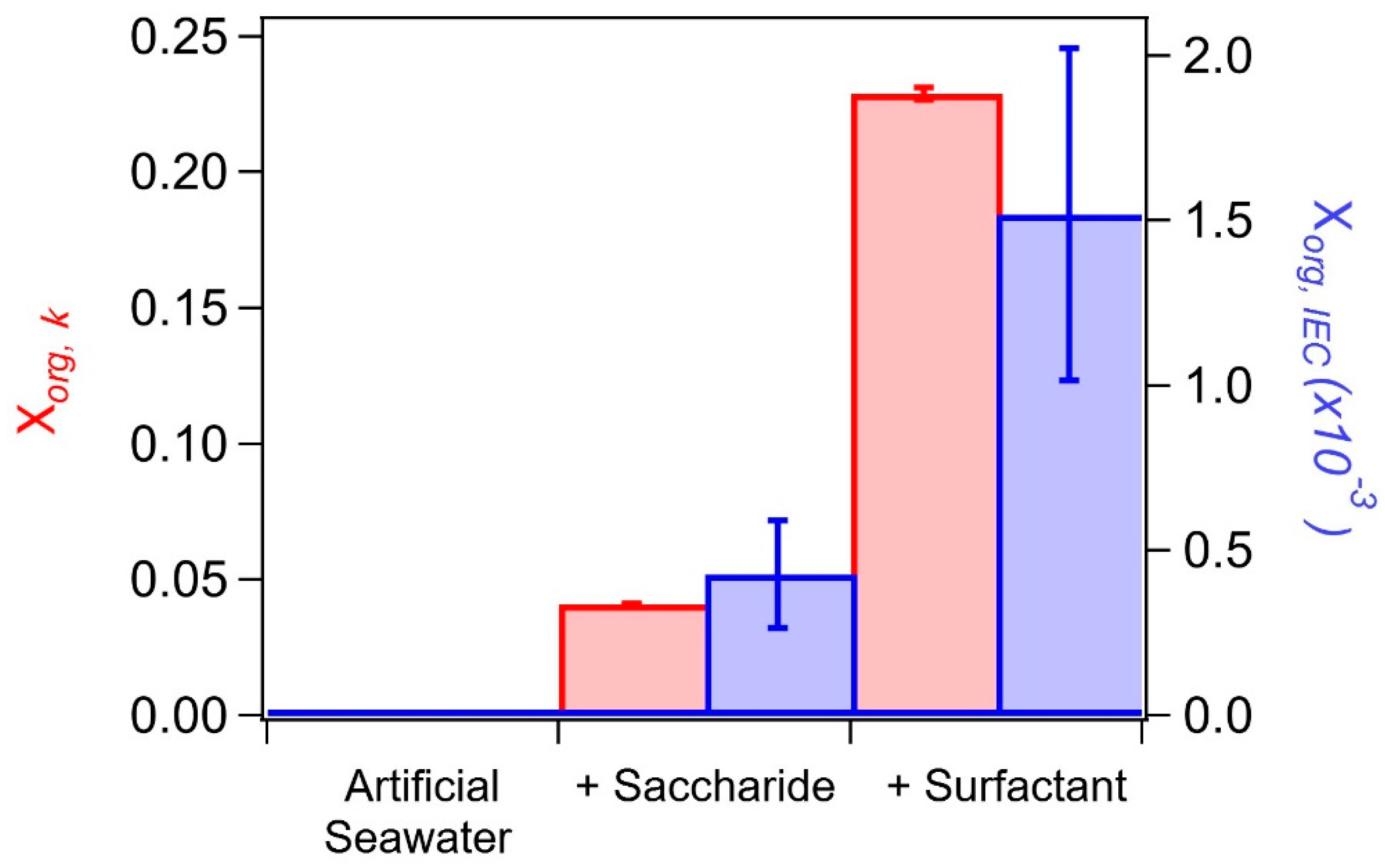
3.3. SSA Organic Enrichment
3.4. Quantitative Measurements of Saccharides in SSA
4. Discussion
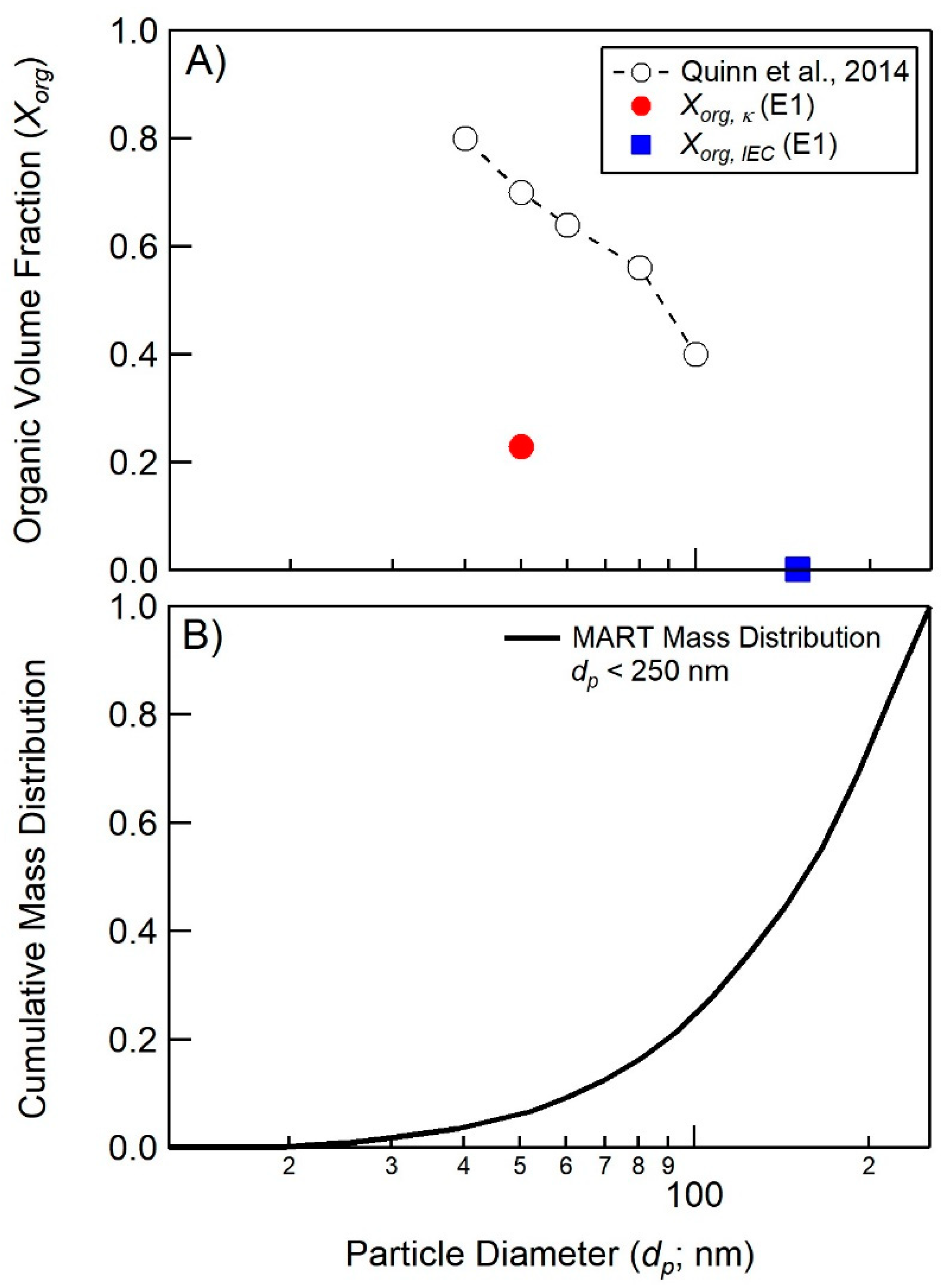
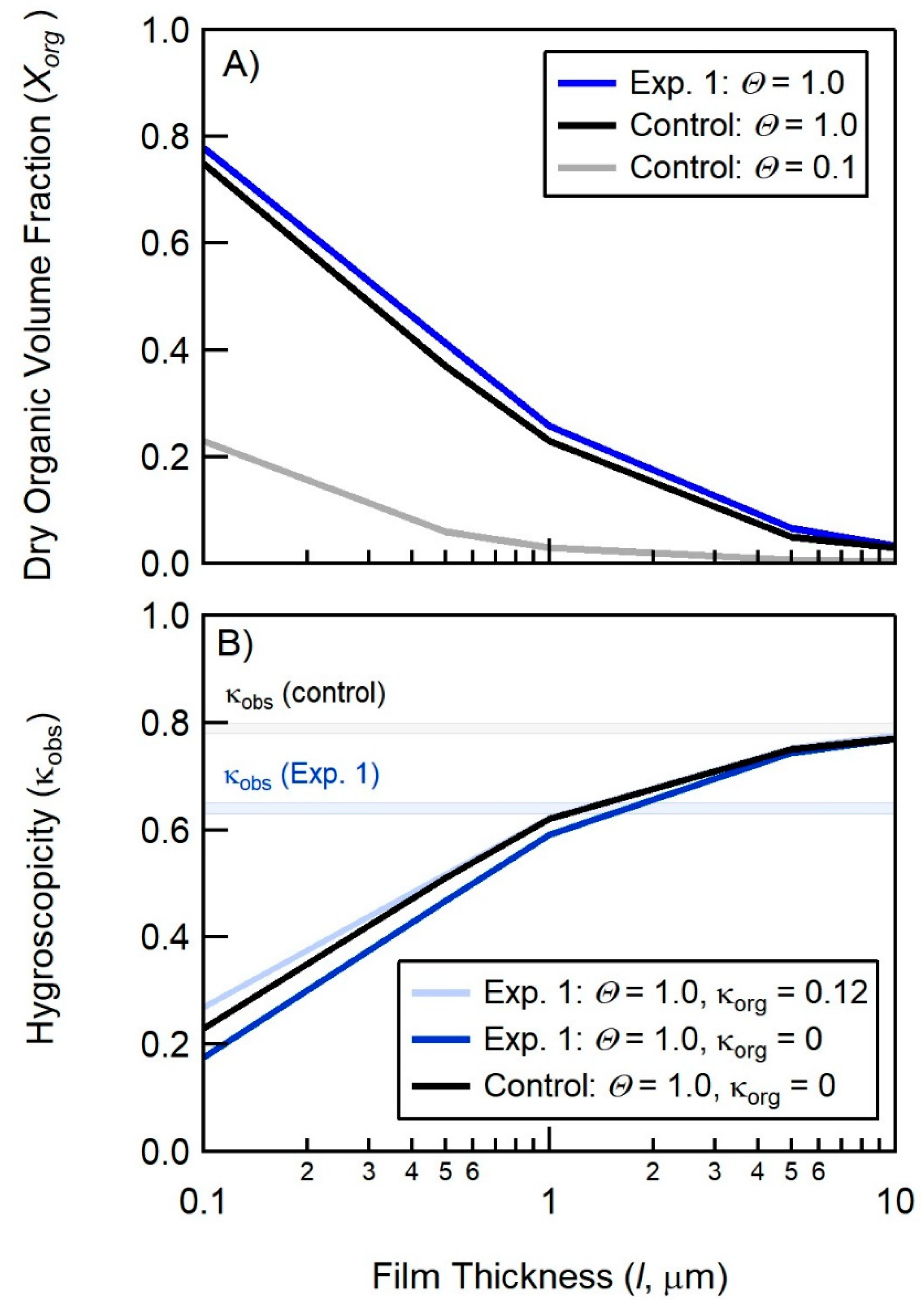
4.1. Comparison of Hygroscopicity and Chromatography Derived SSA Organic Volume Fractions
4.2. Connections between Organic Volume Fractions and SSA Formation Mechanisms
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.-M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and Aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- Aitken, J. XII.—On Dust, Fogs, and Clouds. Earth Environ. Sci. Trans. R. Soc. Edinb. 1881, 30, 337–368. [Google Scholar] [CrossRef]
- Andreae, M.O.; Rosenfeld, D. Aerosol-cloud-precipitation interactions. Part 1. The nature and sources of cloud-active aerosols. Earth Sci. Rev. 2008, 89, 13–41. [Google Scholar] [CrossRef]
- DeMott, P.J.; Hill, T.C.J.; McCluskey, C.S.; Prather, K.A.; Collins, D.B.; Sullivan, R.C.; Ruppel, M.J.; Mason, R.H.; Irish, V.E.; Lee, T.; et al. Sea spray aerosol as a unique source of ice nucleating particles. Proc. Natl. Acad. Sci. USA 2016, 113, 5797–5803. [Google Scholar] [CrossRef] [PubMed]
- Schnell, R.C. Ice Nuclei Produced by Laboratory Cultured Marine Phytoplankton. Geophys. Res. Lett. 1975, 2, 500–502. [Google Scholar] [CrossRef]
- Wilson, T.W.; Ladino, L.A.; Alpert, P.A.; Breckels, M.N.; Brooks, I.M.; Browse, J.; Burrows, S.M.; Carslaw, K.S.; Huffman, J.A.; Judd, C.; et al. A marine biogenic source of atmospheric ice-nucleating particles. Nature 2015, 525, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Ryder, O.S.; Campbell, N.R.; Morris, H.; Forestieri, S.; Ruppel, M.J.; Cappa, C.; Tivanski, A.; Prather, K.; Bertram, T.H. Role of Organic Coatings in Regulating N2O5 Reactive Uptake to Sea Spray Aerosol. J. Phys. Chem. A 2015, 119, 11683–11692. [Google Scholar] [CrossRef]
- Ault, A.P.; Guasco, T.L.; Baltrusaitis, J.; Ryder, O.S.; Trueblood, J.V.; Collins, D.B.; Ruppel, M.J.; Cuadra-Rodriguez, L.A.; Prather, K.A.; Grassian, V.H. Heterogeneous Reactivity of Nitric Acid with Nascent Sea Spray Aerosol: Large Differences Observed between and within Individual Particles. J. Phys. Chem. Lett. 2014, 5, 2493–2500. [Google Scholar] [CrossRef]
- Forestieri, S.D.; Cornwell, G.C.; Helgestad, T.M.; Moore, K.A.; Lee, C.; Novak, G.A.; Sultana, C.M.; Wang, X.; Bertram, T.B.; Prather, K.A.; et al. Linking variations in sea spray aerosol particle hygroscopicity to composition during two microco sm experiments. Atmos. Chem. Phys. 2016, 16, 9003–9018. [Google Scholar] [CrossRef]
- Quinn, P.K.; Bates, T.S.; Schulz, K.S.; Coffman, D.J.; Frossard, A.A.; Russell, L.M.; Keene, W.C.; Kieber, D.J. Contribution of sea surface carbon pool to organic matter enrichment in sea spray aerosol. Nat. Geosci. 2014, 7, 228–232. [Google Scholar] [CrossRef]
- Collins, D.B.; Bertram, T.H.; Sultana, C.M.; Lee, C.; Axson, J.L.; Prather, K.A. Phytoplankton blooms weakly influence the cloud forming ability of sea spray aerosol. Geophys. Res. Lett. 2016, 43, 9975–9983. [Google Scholar] [CrossRef]
- Aluwihare, L.I.; Repeta, D.J.; Chen, R.F. A major biopolymeric component to dissolved organic carbon in surface sea water. Nature 1997, 387, 166–169. [Google Scholar] [CrossRef]
- Hoffman, E.J.; Duce, R.A. Factors Influencing Organic-Carbon Content of Marine Aerosols—Laboratory Study. J. Geophys. Res. Oceans Atmos. 1976, 81, 3667–3670. [Google Scholar] [CrossRef]
- Wang, X.F.; Sultana, C.M.; Trueblood, J.; Hill, T.C.J.; Malfatti, F.; Lee, C.; Laskina, O.; Moore, K.A.; Beall, C.M.; McCluskey, C.S.; et al. Microbial Control of Sea Spray Aerosol Composition: A Tale of Two Blooms. ACS Cent. Sci. 2015, 1, 124–131. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, C.D.; Facchini, M.C.; Cavalli, F.; Ceburnis, D.; Mircea, M.; Decesari, S.; Fuzzi, S.; Yoon, Y.J.; Putaud, J.P. Biogenically driven organic contribution to marine aerosol. Nature 2004, 431, 676–680. [Google Scholar]
- Langmann, B.; Scannell, C.; O’Dowd, C. New Directions: Organic matter contribution to marine aerosols and cloud condensation nuclei. Atmos. Environ. 2008, 42, 7821–7822. [Google Scholar] [CrossRef]
- Mochida, M.; Kitamori, Y.; Kawamura, K. Fatty acids in the marine atmosphere: Factors governing their concentrations and evaluation of organic films on sea-salt particles. J. Geophys. Res. Atmos. 2002, 107, AAC 1-1–AAC 1-10. [Google Scholar] [CrossRef]
- Cochran, R.E.; Jayarathne, T.; Stone, E.A.; Grassian, V.H. Selectivity across the Interface: A Test of Surface Activity in the Composition of Organic-Enriched Aerosols from Bubble Bursting. J. Phys. Chem. Lett. 2016, 7, 1692–1696. [Google Scholar] [CrossRef]
- Jayarathne, T.; Sultana, C.M.; Lee, C.; Malfatti, F.; Cox, J.L.; Pendergraft, M.A.; Moore, K.A.; Azam, F.; Tivanski, A.V.; Cappa, C.D.; et al. Enrichment of Saccharides and Divalent Cations in Sea Spray Aerosol During Two Phytoplankton Blooms. Environ. Sci. Technol. 2016, 50, 11511–11520. [Google Scholar] [CrossRef]
- O’Dowd, C.D.; De Leeuw, G. Marine aerosol production: A review of the current knowledge. Phil. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2007, 365, 1753–1774. [Google Scholar] [CrossRef]
- Keene, W.C.; Maring, H.; Maben, J.R.; Kieber, D.J.; Pszenny, A.A.P.; Dahl, E.E.; Izaguirre, M.A.; Davis, A.J.; Long, M.S.; Zhou, X.; et al. Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface. J. Geophys. Res. Atmos. 2007, 112. [Google Scholar] [CrossRef]
- Barker, D.R.; Zeitlin, H. Metal-Ion Concentrations in Sea-Surface Microlayer and Size-Separated Atmospheric Aerosol Samples in Hawaii. J. Geophys. Res. 1972, 77, 5076–5086. [Google Scholar] [CrossRef]
- Holland, H.D. The Chemistry of Atmosphere and Oceans; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1978. [Google Scholar]
- Sarmiento, J.L.; Gruber, N. Ocean Biogeochemical Dynamics; Princeton University Press: Princeton, NJ, USA, 2006. [Google Scholar]
- Lee, C.; Sultana, C.M.; Collins, D.B.; Santander, M.V.; Axson, J.L.; Malfatti, F.; Cornwell, G.C.; Grandquist, J.R.; Deane, G.B.; Stokes, M.D.; et al. Advancing Model Systems for Fundamental Laboratory Studies of Sea Spray Aerosol Using the Microbial Loop. J. Phys. Chem. A 2015, 119, 8860–8870. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.D.; Hedges, J.I.; Benner, R. Major bacterial contribution to marine dissolved organic nitrogen. Science 1998, 281, 231–234. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Aristegui, J.; Duarte, C.M.; Agusti, S.; Doval, M.; Alvarez-Salgadi, X.A.; Hansell, D.A. Dissolved organic carbon support of respiration in the dark ocean. Science 2002, 298, 1967. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, E.T.; Hayward, N.A. Spatial and temporal variability of total organic carbon along 140 degrees W in the equatorial Pacific Ocean in 1992. Deep Sea Res. Part II Top. Stud. Oceanogr. 1996, 43, 1155–1180. [Google Scholar] [CrossRef]
- Copinmontegut, G.; Avril, B. Vertical-Distribution and Temporal Variation of Dissolved Organic-Carbon in the North-Western Mediterranean-Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 1993, 40, 1963–1972. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Suzuki, Y.; Garside, C.; Ducklow, H.W. High Turnover Rates of Dissolved Organic-Carbon during a Spring Phytoplankton Bloom. Nature 1991, 352, 612–614. [Google Scholar] [CrossRef]
- Norrman, B.; Zweifel, U.L. Production and Utilization of Dissolved Organic-Carbon during an Experimental Diatom Bloom. Limnol. Oceanogr. 1995, 40, 898–907. [Google Scholar] [CrossRef]
- Gobler, C.J.; Sanudo-Wilhelmy, S.A. Cycling of colloidal organic carbon and nitrogen during an estuarine phytoplankton bloom. Limnol. Oceanogr. 2003, 48, 2314–2320. [Google Scholar] [CrossRef]
- Facchini, M.C.; Rinaldi, M.; Decesari, S.; Carbone, C.; Finessi, E.; Mircea, M.; Fuzzi, S.; Ceburnis, D.; Flanagan, R.; Nilsson, E.D.; et al. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef]
- Russell, L.M.; Hawkins, L.N.; Frossard, A.A.; Quinn, P.K.; Bates, T.S. Carbohydrate-like composition of submicron atmospheric particles and their production from ocean bubble bursting. Proc. Natl. Acad. Sci. USA 2010, 107, 6652–6657. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, L.N.; Russell, L. Polysaccharides, Proteins, and Phytoplankton Fragments: Four Chemically Distinct Types of Marine Primary Organic Aerosol Classified by Single Particle Spectromicroscopy. Adv. Meteorol. 2010, 2010, 14. [Google Scholar] [CrossRef]
- Frossard, A.A.; Russell, L.M.; Burrows, S.M.; Elliott, S.M.; Bates, T.S.; Quinn, P.K. Sources and composition of submicron organic mass in marine aerosol particles. J. Geophys. Res. Atmos. 2014, 119, 12977–13003. [Google Scholar] [CrossRef]
- Crahan, K.K.; Hegg, D.A.; Covert, D.S. Speciation of organic aerosols in the tropical mid-pacific and their relationship to light scattering. J. Atmos. Sci. 2004, 61, 2544–2558. [Google Scholar] [CrossRef]
- Aller, J.Y.; Radway, J.C.; Kilthau, W.P.; Bothe, D.W.; Wilson, T.W.; Vailancourt, R.D.; Quinn, P.K.; Coffman, D.J.; Murray, B.J.; Knopf, D.A. Size-resolved characterization of the polysaccharidic and proteinaceous components of sea spray aerosol. Atmos. Environ. 2017, 154, 331–347. [Google Scholar] [CrossRef]
- van Pinxteren, M.; Muller, C.; Iinuma, Y.; Stolle, C.; Herrmann, H. Chemical Characterization of Dissolved Organic Compounds from Coastal Sea Surface Micro layers (Baltic Sea, Germany). Environ. Sci. Technol. 2012, 46, 10455–10462. [Google Scholar] [CrossRef]
- Compiano, A.M.; Romano, J.; de la Giraudiere, I.; Laborde, P. Monosaccharide Composition of Suspended Organic Particulate Matter in Relation to Its Origin. Oceanol. Acta 1993, 16, 135–144. [Google Scholar]
- Compiano, A.M.; Romno, J.; Garabetian, F.; Laborde, P.; de la Giraudierea, I. Monosaccharide Composition of Particulate Hydrolyzable Sugar Fraction in Surface Microlayers from Brackish and Marine Waters. Mar. Chem. 1993, 42, 237–251. [Google Scholar] [CrossRef]
- Haug, A.; Myklestad, S. Polysaccharides of Marine Diatoms with Special Reference to Chaetoceros Species. Mar. Boil. 1976, 34, 217–222. [Google Scholar] [CrossRef]
- Verdugo, P.; Alldredge, A.L.; Azam, F.; Kirchman, D.L.; Passow, U.; Santschi, P.H. The oceanic gel phase: A bridge in the DOM-POM continuum. Mar. Chem. 2004, 92, 67–85. [Google Scholar] [CrossRef]
- Ittekkot, V. Variations of Dissolved Organic-Matter during a Plankton Bloom—Qualitative Aspects, Based on Sugar and Amino-Acid Analyses. Mar. Chem. 1982, 11, 143–158. [Google Scholar] [CrossRef]
- Ittekkot, V.; Degens, E.T.; Brockmann, U. Monosaccharide Composition of Acid-Hydrolyzable Carbohydrates in Particulate Matter during a Plankton Bloom. Limnol. Oceanogr. 1982, 27, 770–776. [Google Scholar] [CrossRef]
- Liebezeit, G.; Bolter, M.; Brown, I.F.; Dawson, R. Dissolved Free Amino-Acids and Carbohydrates at Pycnocline Boundaries in the Sargasso Sea and Related Microbial Activity. Oceanol. Acta 1980, 3, 357–362. [Google Scholar]
- Pakulski, J.D.; Benner, R. An Improved Method for the Hydrolysis and Mbth Analysis of Dissolved and Particulate Carbohydrates in Seawater. Mar. Chem. 1992, 40, 143–160. [Google Scholar] [CrossRef]
- Benner, R.; Pakulski, J.D.; McCarthy, M.; Hedges, J.I.; Hatcher, P.G. Bulk Chemical Characteristics of Dissolved Organic-Matter in the Ocean. Science 1992, 255, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Mopper, K. Sugars and Uronic-Acids in Sediment and Water from Black Sea and North-Sea with Emphasis on Analytical Techniques. Mar. Chem. 1977, 5, 585–603. [Google Scholar] [CrossRef]
- Sakugawa, H.; Handa, N. Isolation and Chemical Characterization of Dissolved and Particulate Polysaccharides in Mikawa Bay. Geochim. Cosmochim. Acta 1985, 49, 1185–1193. [Google Scholar] [CrossRef]
- Mopper, K.; Zhou, J.; Ramana, K.S.; Passow, U.; Dam, H.G.; Drapeau, D.T. The Role of Surface-Active Carbohydrates in the Flocculation of a Diatom Bloom in a Mesocosm. Deep Sea Res. Part II Top. Stud. Oceanogr. 1995, 42, 47–73. [Google Scholar] [CrossRef]
- Gao, Q.; Leck, C.; Rauschenberg, C.; Matrai, P.A. On the chemical dynamics of extracellular polysaccharides in the high Arctic surface microlayer. Ocean Sci. 2012, 8, 401–418. [Google Scholar] [CrossRef]
- Zhou, J.; Mopper, K.; Passow, U. The role of surface-active carbohydrates in the formation of transparent exopolymer particles by bubble adsorption of seawater. Limnol. Oceanogr. 1998, 43, 1860–1871. [Google Scholar] [CrossRef]
- Cochran, R.E.; Laskina, O.; Jayarathne, T.; Laskin, A.; Laskin, J.; Lin, P.; Sultana, C.; Lee, C.; Moore, K.A.; Cappa, C.D.; et al. Analysis of Organic Anionic Surfactants in Fine and Coarse Fractions of Freshly Emitted Sea Spray Aerosol. Environ. Sci. Technol. 2016, 50, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Burrows, S.M.; Ogunro, O.; Frossard, A.A.; Russell, L.M.; Rasch, P.J.; Elliott, S.M. A physically based framework for modeling the organic fractionation of sea spray aerosol from bubble film Langmuir equilibria. Atmos. Chem. Phys. 2014, 14, 13601–13629. [Google Scholar] [CrossRef]
- Burrows, S.M.; Gobrogge, E.; Fu, L.; Link, K.; Elliott, S.M.; Wang, H.; Walker, R. OCEANFILMS-2: Representing coadsorption of saccharides in marine films and potential impacts on modeled marine aerosol chemistry. Geophys. Res. Lett. 2016, 43, 8306–8313. [Google Scholar] [CrossRef]
- Nakahara, H.; Lee, S.; Shoyama, Y.; Shibata, O. The role of palmitic acid in pulmonary surfactant systems by Langmuir monolayer study: Lipid-peptide interactions. Soft Matter 2011, 7, 11351–11359. [Google Scholar] [CrossRef]
- Papahadjopoulos, D. Surface Properties of Acidic Phospholipids—Interaction of Monolayers and Hydrated Liquid Crystals with Uni-and Bi-Valent Metal Ions. Biochim. Biophys. Acta 1968, 163, 240–254. [Google Scholar] [CrossRef]
- Bichsel, Y.; Von Gunten, U. Formation of iodo-trihalomethanes during disinfection and oxidation of iodide containing waters. Environ. Sci. Technol. 2000, 34, 2784–2791. [Google Scholar] [CrossRef]
- Pörtner, H.-O.; Karl, D.M.; Boyd, P.W.; Cheung, W.W.L.; Lluch-Cota, S.E.; Nojiri, Y.; Schmidt, D.N.; Zavialov, P.O. Ocean systems. In Climate Change 2014: Impacts, Adaptation, and Vulnerability. Part A: Global and Sectoral Aspects. Contribution of Working Group II to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2014; pp. 411–484. [Google Scholar]
- Quinn, P.K.; Collins, D.B.; Grassian, V.H.; Prather, K.A.; Bates, T.S. Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol. Chem. Rev. 2015, 115, 4383–4399. [Google Scholar] [CrossRef] [PubMed]
- Kanicky, J.R.; Poniatowski, A.F.; Mehta, N.R.; Shah, D.O. Cooperativity among molecules at interfaces in relation to various technological processes: Effect of chain length on the pKa of fatty acid salt solutions. Langmuir 2000, 16, 172–177. [Google Scholar] [CrossRef]
- Wang, H.M.; Loganathan, D.; Linhardt, R.J. Determination of the Pka of Glucuronic-Acid and the Carboxy Groups of Heparin by C-13-Nuclear-Magnetic-Resonance Spectroscopy. Biochem. J. 1991, 278, 689–695. [Google Scholar] [CrossRef]
- Tang, C.Y.; Huang, Z.S.A.; Allen, H.C. Binding of Mg2+ and Ca2+ to Palmitic Acid and Deprotonation of the COOH Headgroup Studied by Vibrational Sum Frequency Generation Spectroscopy. J. Phys. Chem. B 2010, 114, 17068–17076. [Google Scholar] [CrossRef] [PubMed]
- Wellen Rudd, B.A.; Vidalis, A.S.; Allen, H.C. Thermodynamic versus Non-Equilibrium Stability of Palmitic Acid Monolayers in Calcium-Enriched Sea Spray Aerosol Proxy Systems. Phys. Chem. Chem. Phys. 2018, 20, 16320–16332. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.D.; Deane, G.B.; Prather, K.; Bertram, T.H.; Ruppel, M.J.; Ryder, O.S.; Brady, J.M.; Zhao, D. A Marine Aerosol Reference Tank system as a breaking wave analogue for the production of foam and sea-spray aerosols. Atmos. Meas. Tech. 2013, 6, 1085–1094. [Google Scholar] [CrossRef]
- Schill, S.R.; Collins, D.B.; Lee, C.; Morris, H.S.; Novak, G.A.; Prather, K.A.; Quinn, P.K.; Sultana, C.M.; Tivanski, A.V.; Zimmermann, K.; et al. The Impact of Aerosol Particle Mixing State on the Hygroscopicity of Sea Spray Aerosol. ACS Cent. Sci. 2015, 1, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, L.; Rohrer, J.S. Applications of Ion Chromatography for Pharmaceutical and Biological Products; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Hegg, D.A.; Covert, D.S.; Jonsson, H.H.; Woods, R.K. A simple relationship between cloud drop number concentration and precursor aerosol concentration for the regions of Earth’s large marine stratocumulus decks. Atmos. Chem. Phys. 2012, 12, 1229–1238. [Google Scholar] [CrossRef]
- Farmer, D.K.; Cappa, C.D.; Kreidenweis, S.M. Atmospheric Processes and Their Controlling Influence on Cloud Condensation Nuclei Activity. Chem. Rev. 2015, 115, 4199–4217. [Google Scholar] [CrossRef]
- Petters, M.D.; Kreidenweis, S.M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961–1971. [Google Scholar] [CrossRef]
- Stokes, R.H.; Robinson, R.A. Interactions in aqueous nonelectrolyte solutions. I. Solute-solvent equilibria. J. Phys. Chem. 1966, 70, 2126–2131. [Google Scholar] [CrossRef]
- Dawson, K.W.; Petters, M.D.; Meskhidze, N.; Suda Petters, S.; Kreidenweis, S.M. Hygroscopic growth and cloud droplet activation of xanthan gum as a proxy for marine hydrogels. J. Geophys. Res. Atmos. 2016, 121, 11803–11818. [Google Scholar] [CrossRef]
- Bertram, T.H.; Cochran, R.E.; Grassian, V.H.; Stone, E.A. Sea spray aerosol chemical composition: Elemental and molecular mimics for laboratory studies of heterogeneous and multiphase reactions. Chem. Soc. Rev. 2018, 47, 2374–2400. [Google Scholar] [CrossRef]
- Zobrist, B.; Soonsin, V.; Luo, B.P.; Krieger, U.K.; Marcolli, C.; Peter, T.; Koop, T. Ultra-slow water diffusion in aqueous sucrose glasses. Phys. Chem. Chem. Phys. 2011, 13, 3514–3526. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.J.; Reid, J.P.; Bones, D.L.; Luo, B.P.; Krieger, U.K. Measurements of the timescales for the mass transfer of water in glassy aerosol at low relative humidity and ambient temperature. Atmos. Chem. Phys. 2011, 11, 4739–4754. [Google Scholar] [CrossRef]
- Prather, K.A.; Bertram, T.H.; Grassian, V.H.; Deane, G.B.; Stokes, M.D.; DeMott, P.J.; Aluwihare, L.I.; Palenik, B.P.; Azam, F.; Seinfeld, J.H.; et al. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol. Proc. Natl. Acad. Sci. USA 2013, 110, 7550–7555. [Google Scholar] [CrossRef] [PubMed]
- Modini, R.L.; Russell, L.M.; Deane, G.B.; Stokes, M.D. Effect of soluble surfactant on bubble persistence and bubble-produced aerosol particles. J. Geophys. Res. Atmos. 2013, 118, 1388–1400. [Google Scholar] [CrossRef]
- Garrett, W.D. Stabilization of air bubbles at the air-sea interface by surface-active material. Deep Sea Res. Oceanogr. Abstr. 1967, 14, 661–672. [Google Scholar] [CrossRef]
- Ternes, R.L.; Berg, J.C. The effect of monolayer collapse on bubble stability. J. Colloid Interface Sci. 1984, 98, 471–477. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Artificial Salt Water | + Saccharide † | + Surfactant † | |||||
|---|---|---|---|---|---|---|---|
| Na+ (mM) | Mg2+ (mM) | Ca2+ (mM) | Cl− (mM) | + Glucose (μM) | + Glucuronic Acid (μM) | + Palmitic Acid (μM) | |
| Control | 470 | 145 | 0 | 600 | 95 | 0 | 35 |
| E1 | 470 | 145 | 0 | 600 | 0 | 95 | 35 |
| E2 | 470 | 0 | 10 | 490 | 0 | 95 | 35 |
| E3 | 470 | 0 | 0 | 470 | 0 | 95 | 35 |
| Artificial Seawater | + Saccharide † | +Surfactant † | |||||
|---|---|---|---|---|---|---|---|
| κ | κ | Δ | Xorg, κ | κ | Δ | XXorg, κ | |
| Control | 0.749 ± 0.010 | 0.744 ± 0.002 | −1% | 0.007 | 0.734 ± 0.001 | −2% | 0.022 |
| E1 | 0.797 ± 0.005 | 0.767 ± 0.004 | −4% | 0.041 | 0.630 ± 0.009 | −21% | 0.229 |
| E2 | 0.921 ± 0.013 | 0.907 ± 0.014 | −2% | 0.016 | 0.646 ± 0.006 | −30% | 0.323 |
| E3 | 0.892 ± 0.005 | 0.889 ± 0.026 | <−1% | 0.004 | 0.773 ± 0.013 | −13% | 0.144 |
| Artificial Seawater | + Saccharide | + Surfactant | |
|---|---|---|---|
| Xsaccharide, IEC | Xsaccharide, IEC | Xsaccharide, IEC | |
| Control | 2.7 × 10−3 ± 7.3 × 10−4 | 3.1 × 10−3 ± 8.4 × 10−4 | 2.4 × 10−3 ± 7.9 × 10−4 |
| E1 | ND † | 1.2 × 10−4 ± 4.6 × 10−5 | 4.3 × 10−4 ± 1.4 × 10−4 |
| E2 | ND † | 1.7 × 10−4 ± 5.4 × 10−5 | 4.9 × 10−4 ± 1.6 × 10−4 |
| E3 | ND † | 5.5 × 10−4 ± 1.5 × 10−4 | 7.4 × 10−4 ± 2.4 × 10−4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schill, S.R.; Burrows, S.M.; Hasenecz, E.S.; Stone, E.A.; Bertram, T.H. The Impact of Divalent Cations on the Enrichment of Soluble Saccharides in Primary Sea Spray Aerosol. Atmosphere 2018, 9, 476. https://doi.org/10.3390/atmos9120476
Schill SR, Burrows SM, Hasenecz ES, Stone EA, Bertram TH. The Impact of Divalent Cations on the Enrichment of Soluble Saccharides in Primary Sea Spray Aerosol. Atmosphere. 2018; 9(12):476. https://doi.org/10.3390/atmos9120476
Chicago/Turabian StyleSchill, Steven R., Susannah M. Burrows, Elias S. Hasenecz, Elizabeth A. Stone, and Timothy H. Bertram. 2018. "The Impact of Divalent Cations on the Enrichment of Soluble Saccharides in Primary Sea Spray Aerosol" Atmosphere 9, no. 12: 476. https://doi.org/10.3390/atmos9120476
APA StyleSchill, S. R., Burrows, S. M., Hasenecz, E. S., Stone, E. A., & Bertram, T. H. (2018). The Impact of Divalent Cations on the Enrichment of Soluble Saccharides in Primary Sea Spray Aerosol. Atmosphere, 9(12), 476. https://doi.org/10.3390/atmos9120476