Monthly and Diurnal Variation of the Concentrations of Aerosol Surface Area in Fukuoka, Japan, Measured by Diffusion Charging Method


Abstract
:1. Introduction
2. Experiments
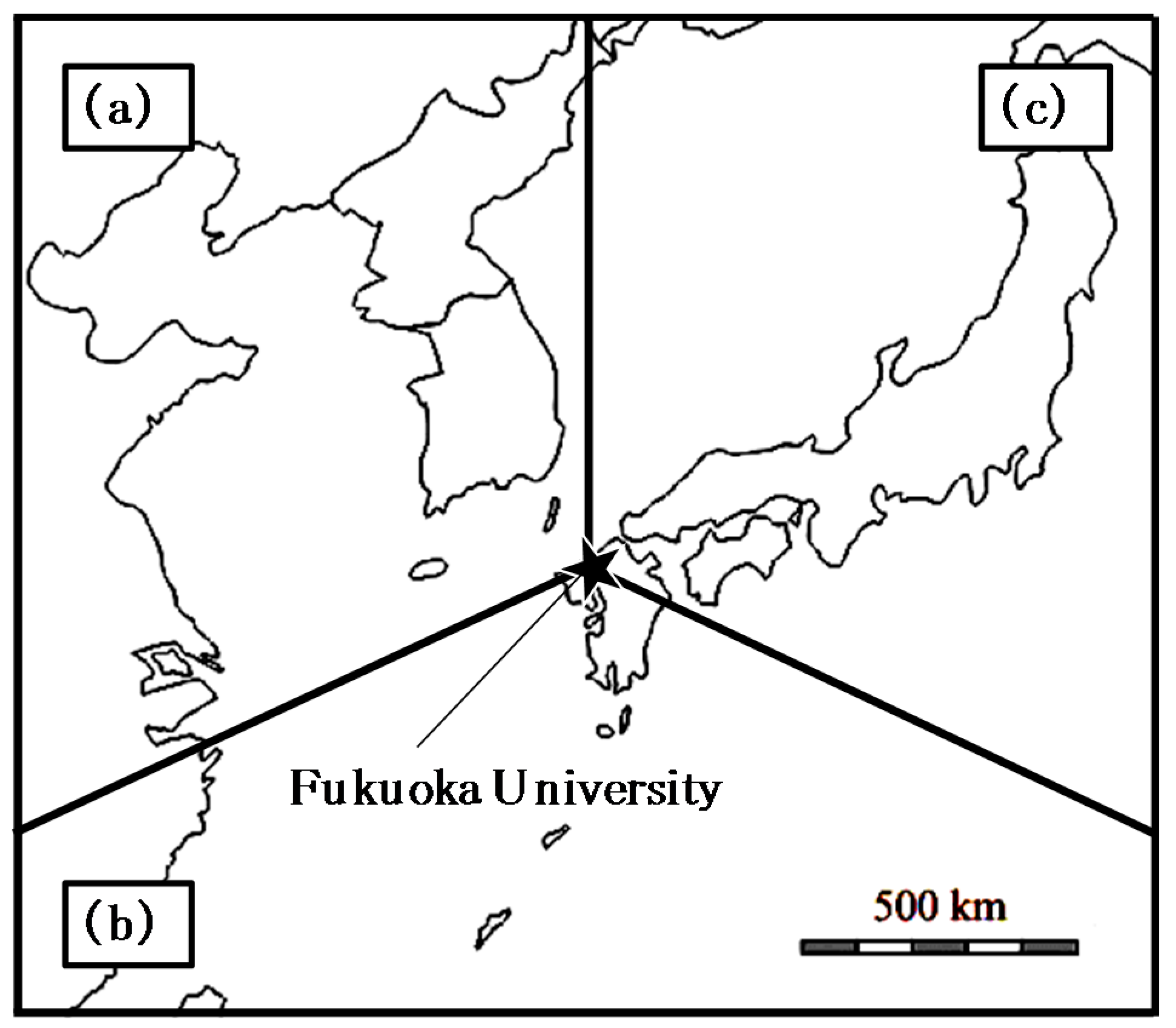
2.1. Observation Site and Period
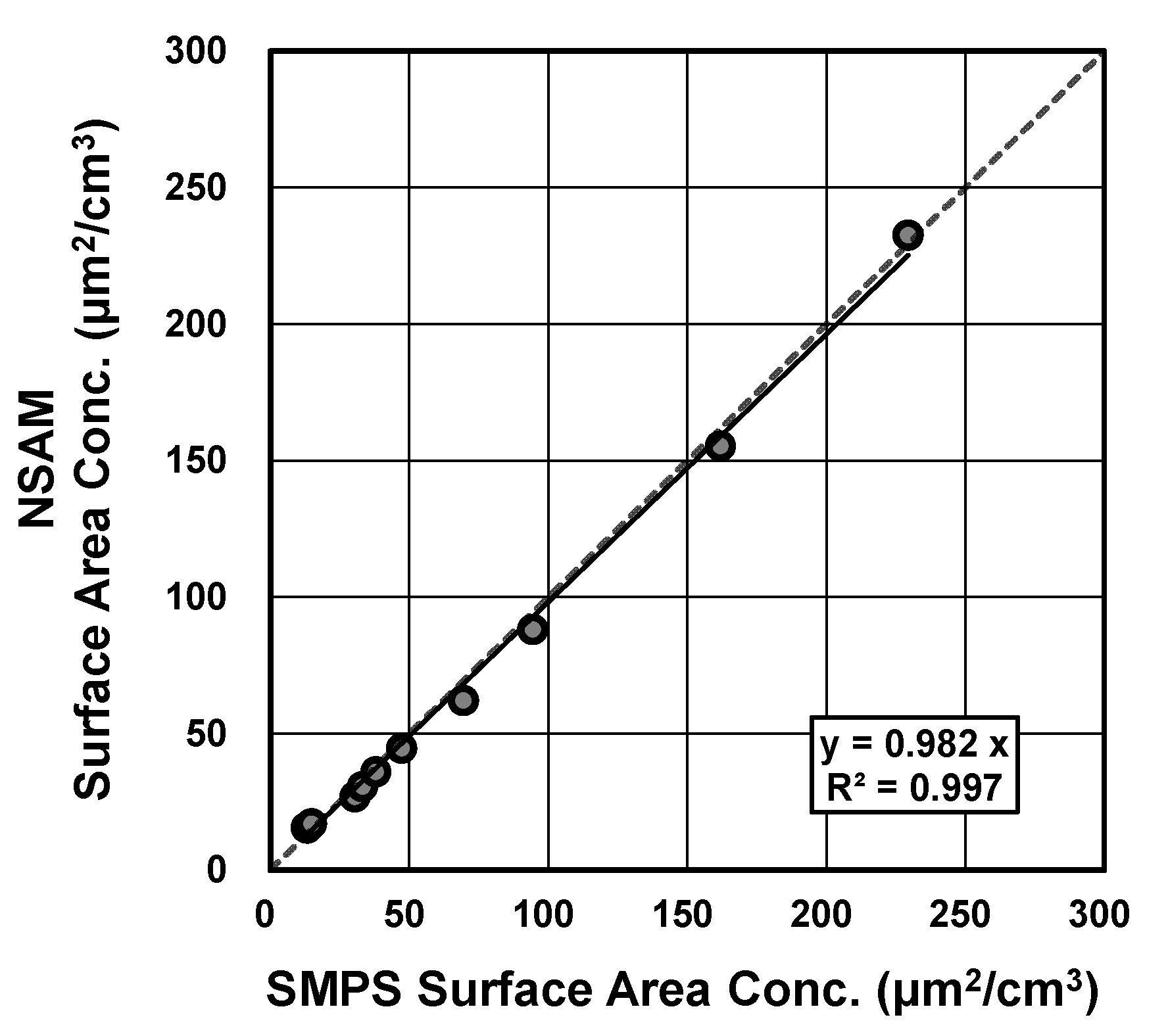
2.2. Aerosol Surface Area
2.3. Black Carbon, Aerosol Number and Mass, Sulfate Ion Concentration, and Wind Direction and Speed
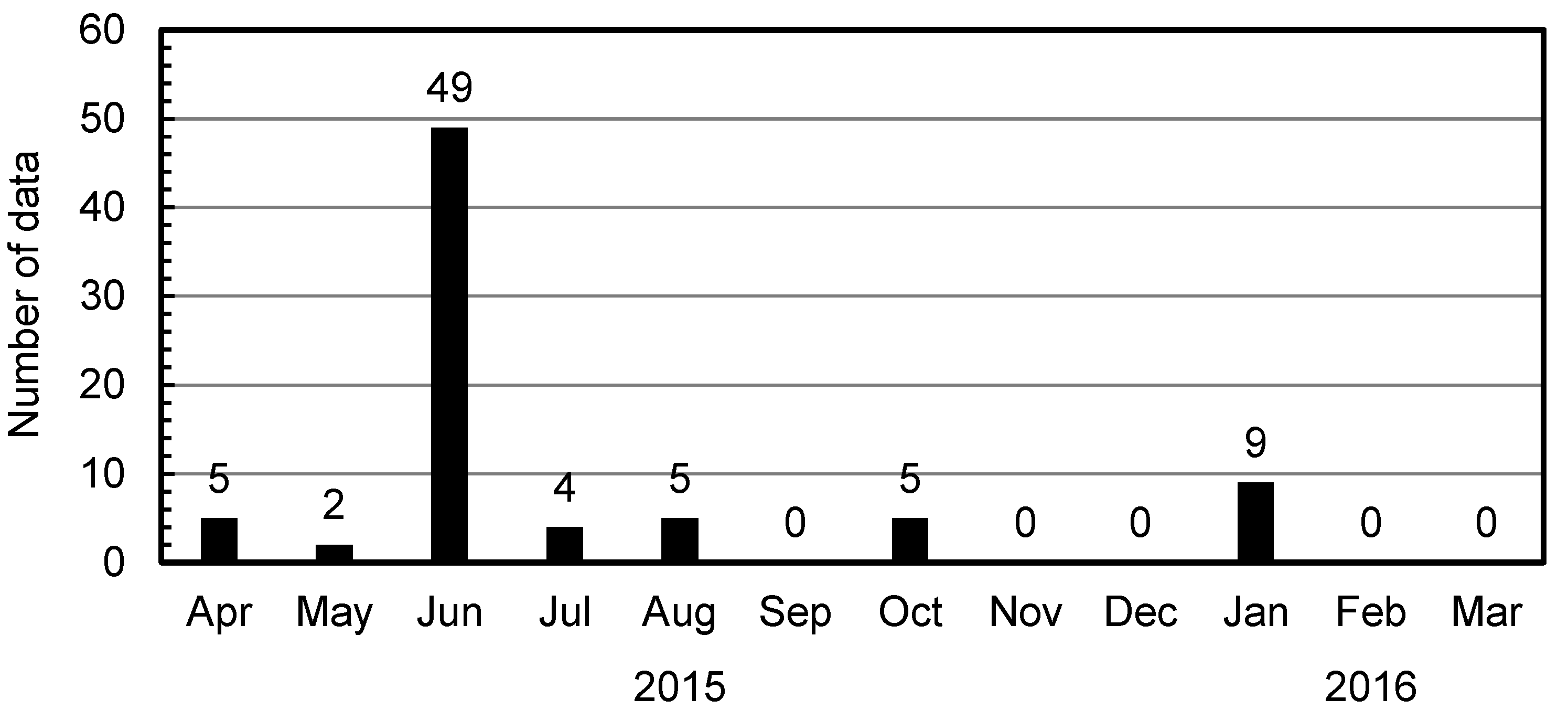
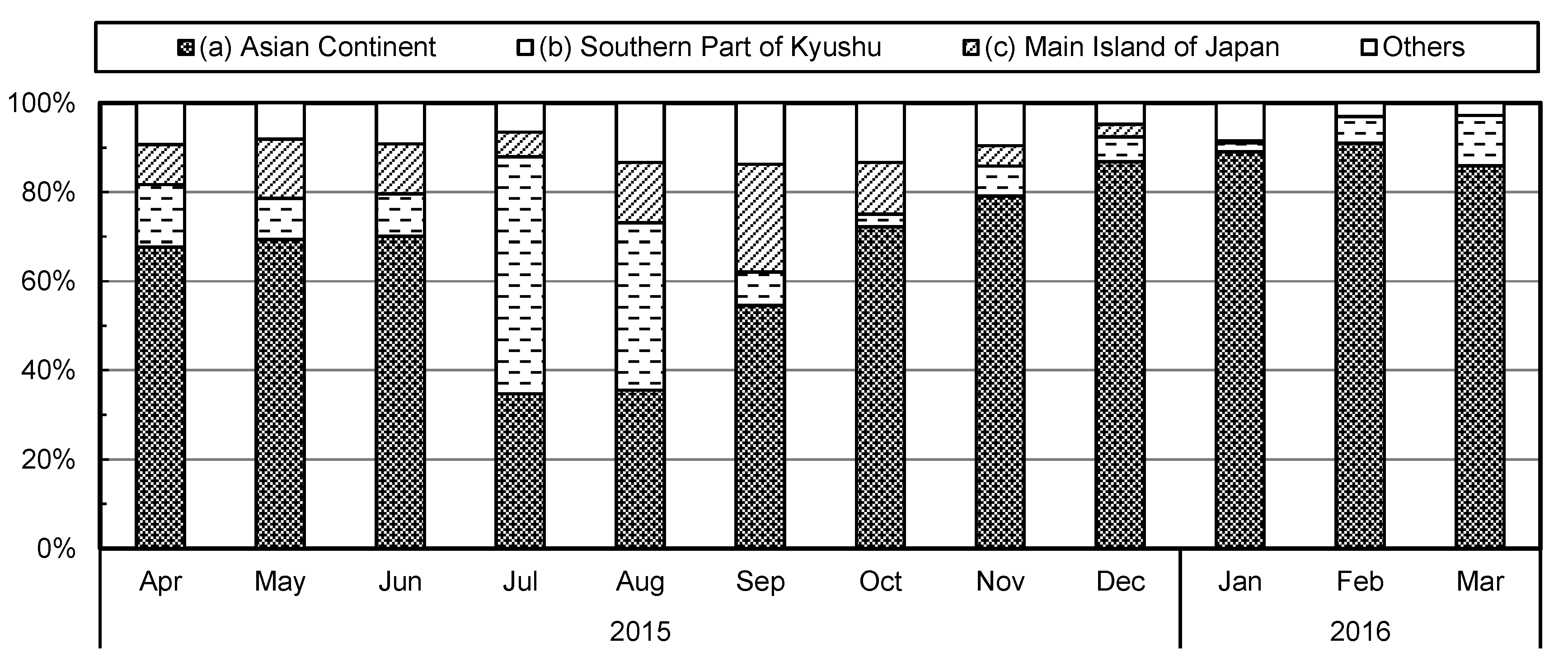
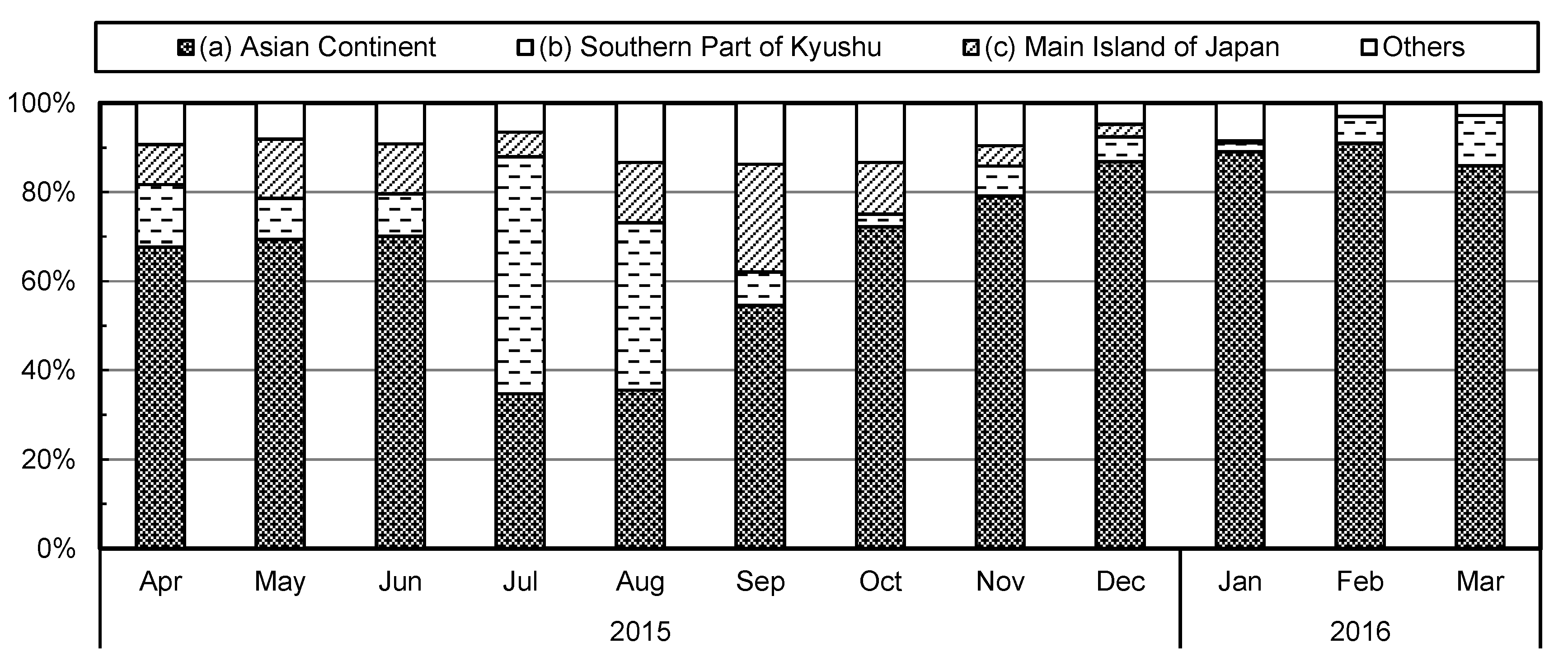
2.4. Airmass Backward Trajectory Analysis
3. Results and Discussion
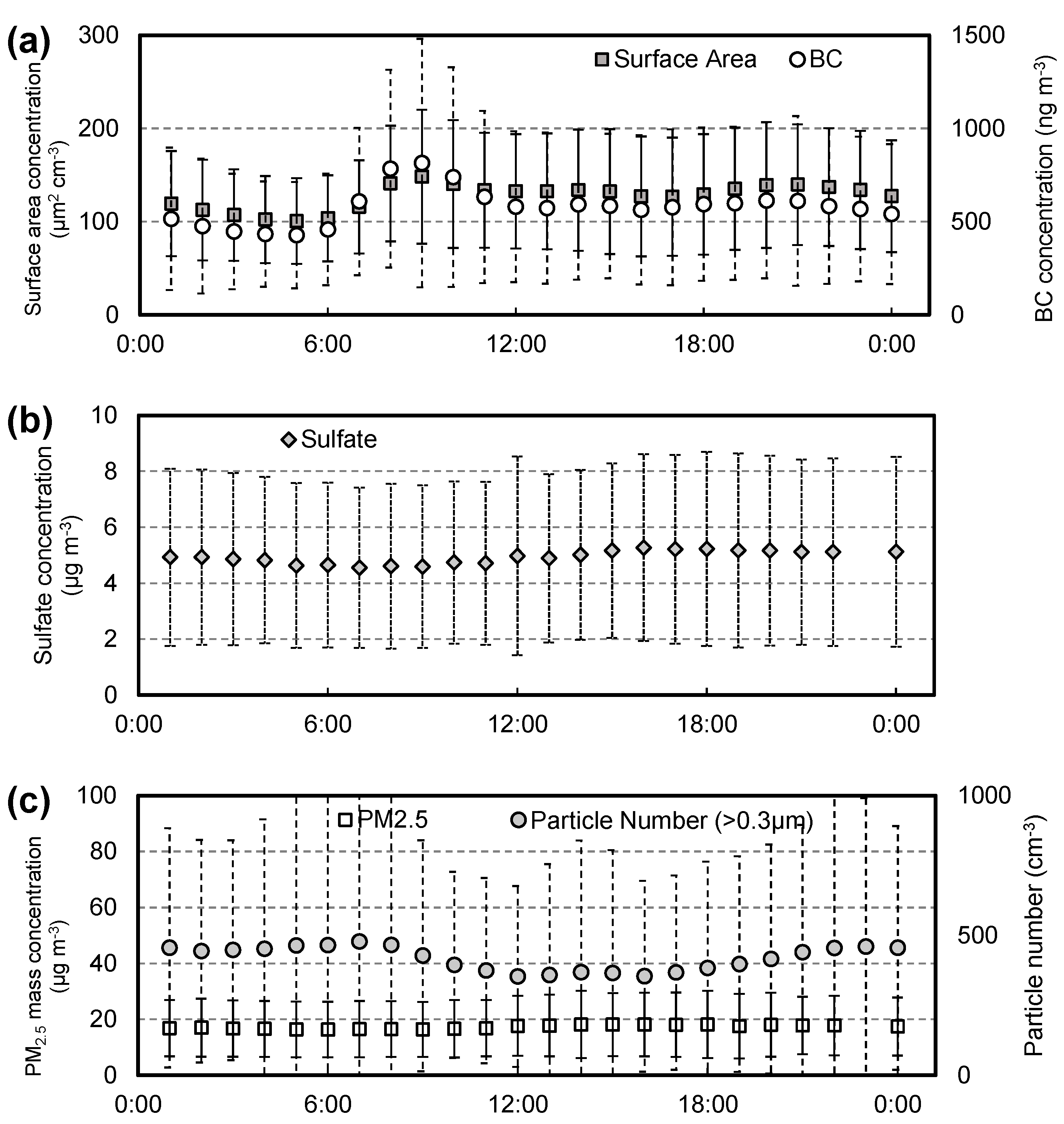
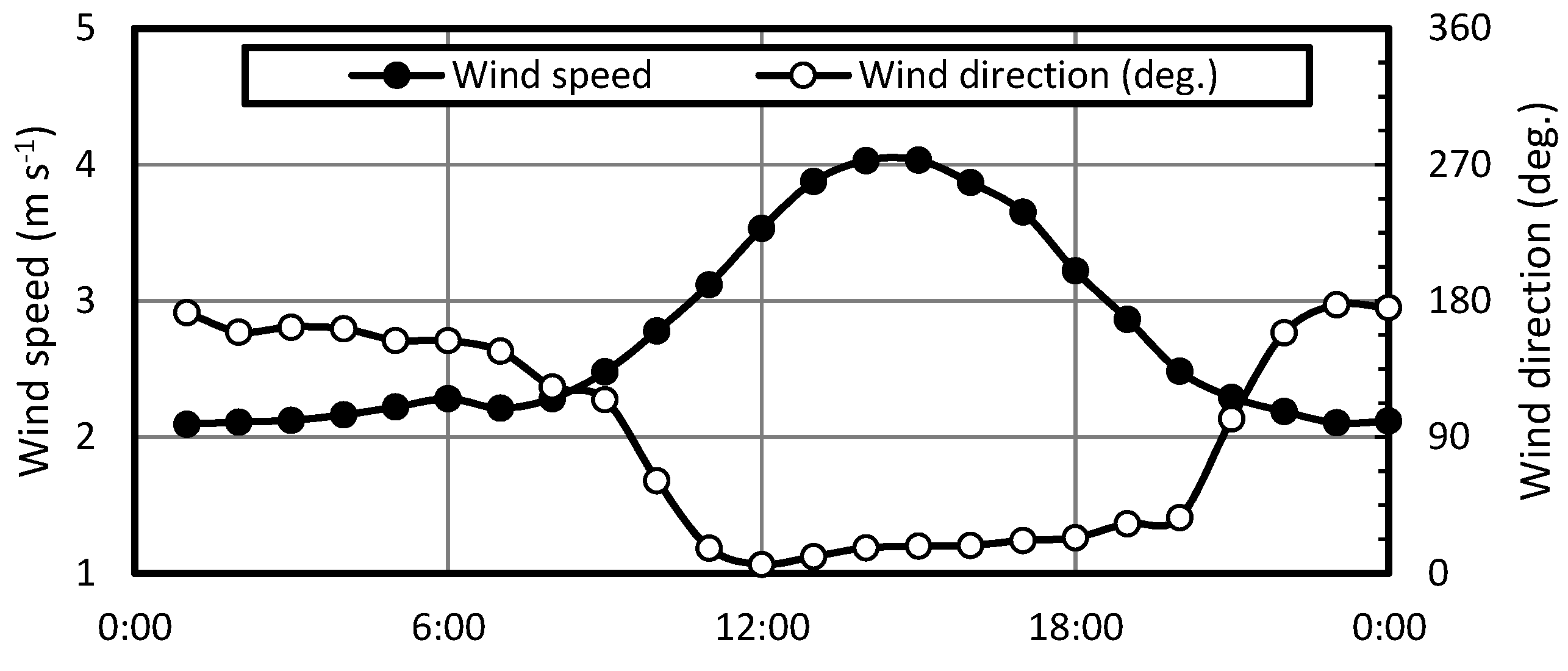
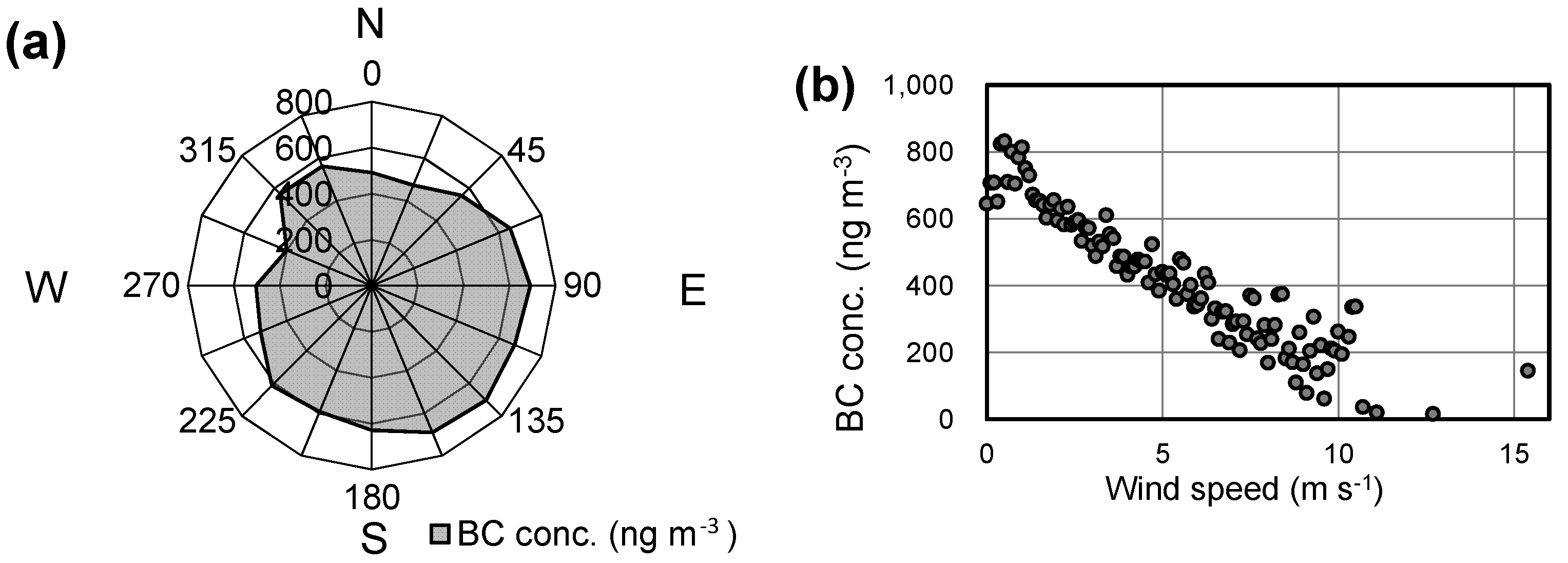
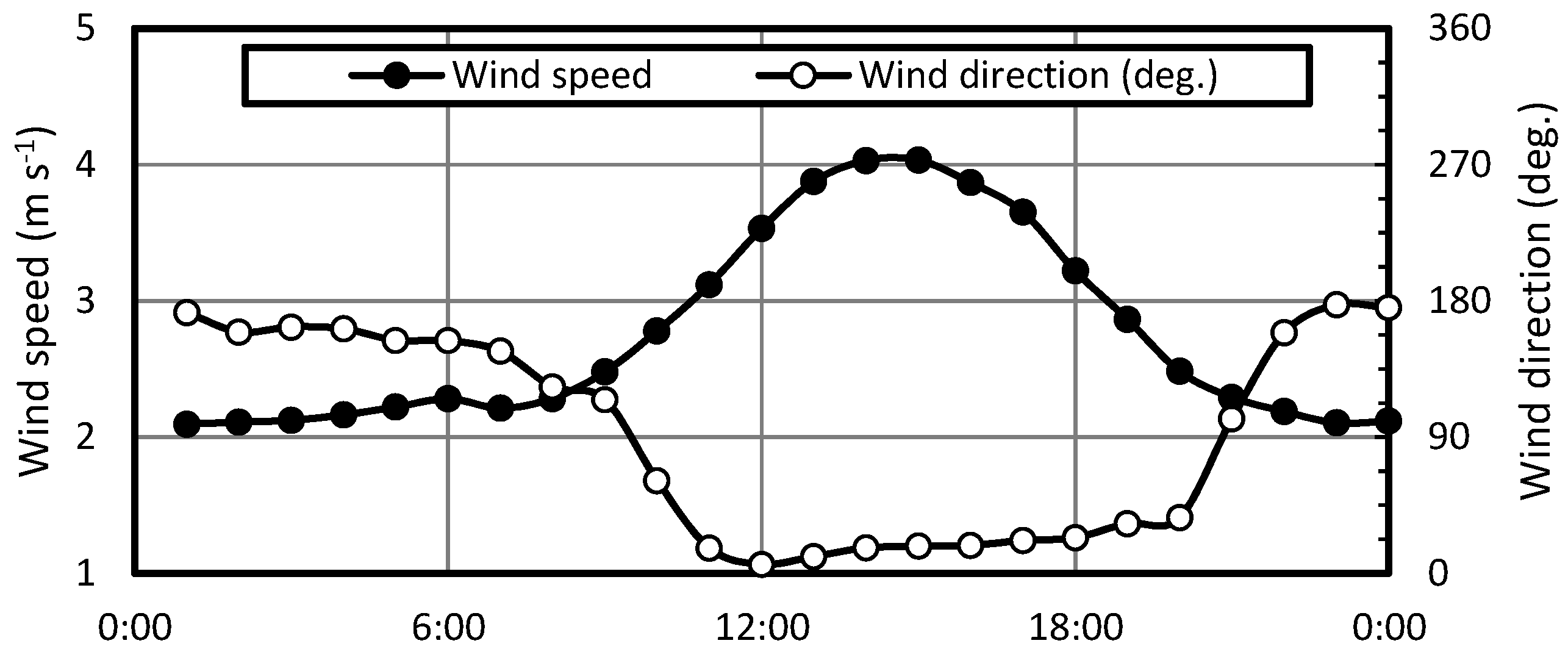
3.1. Diurnal Variations
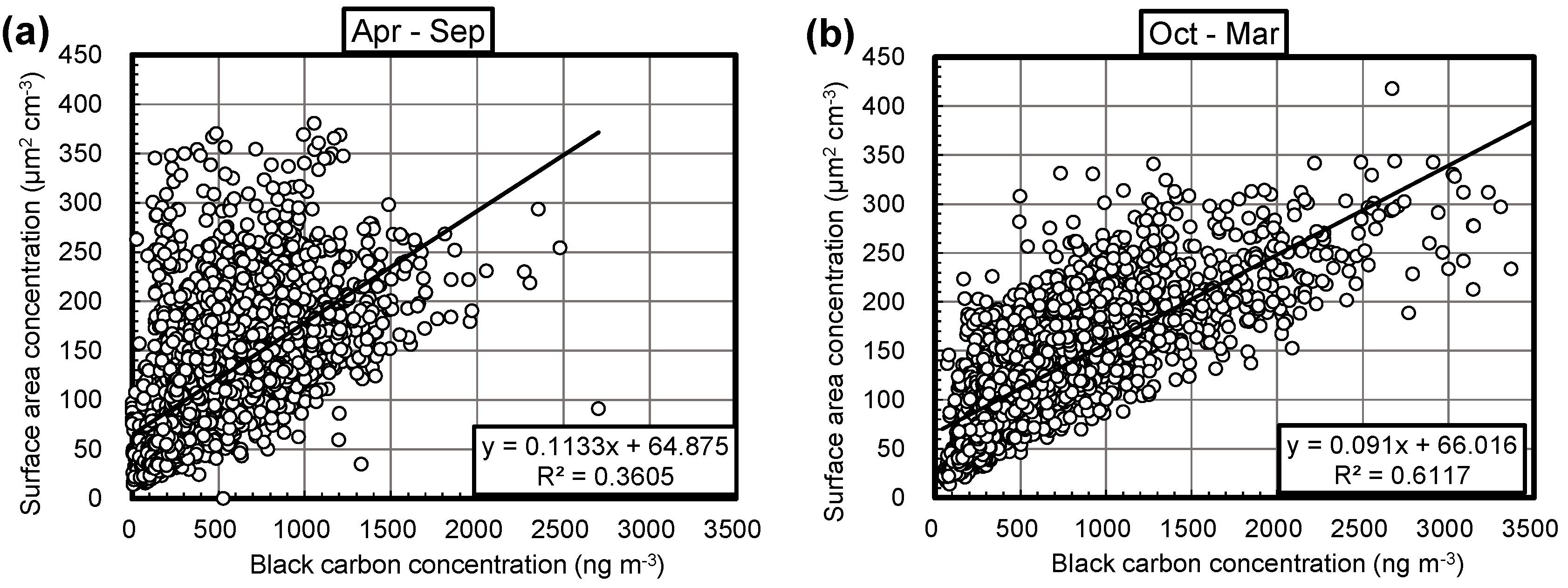
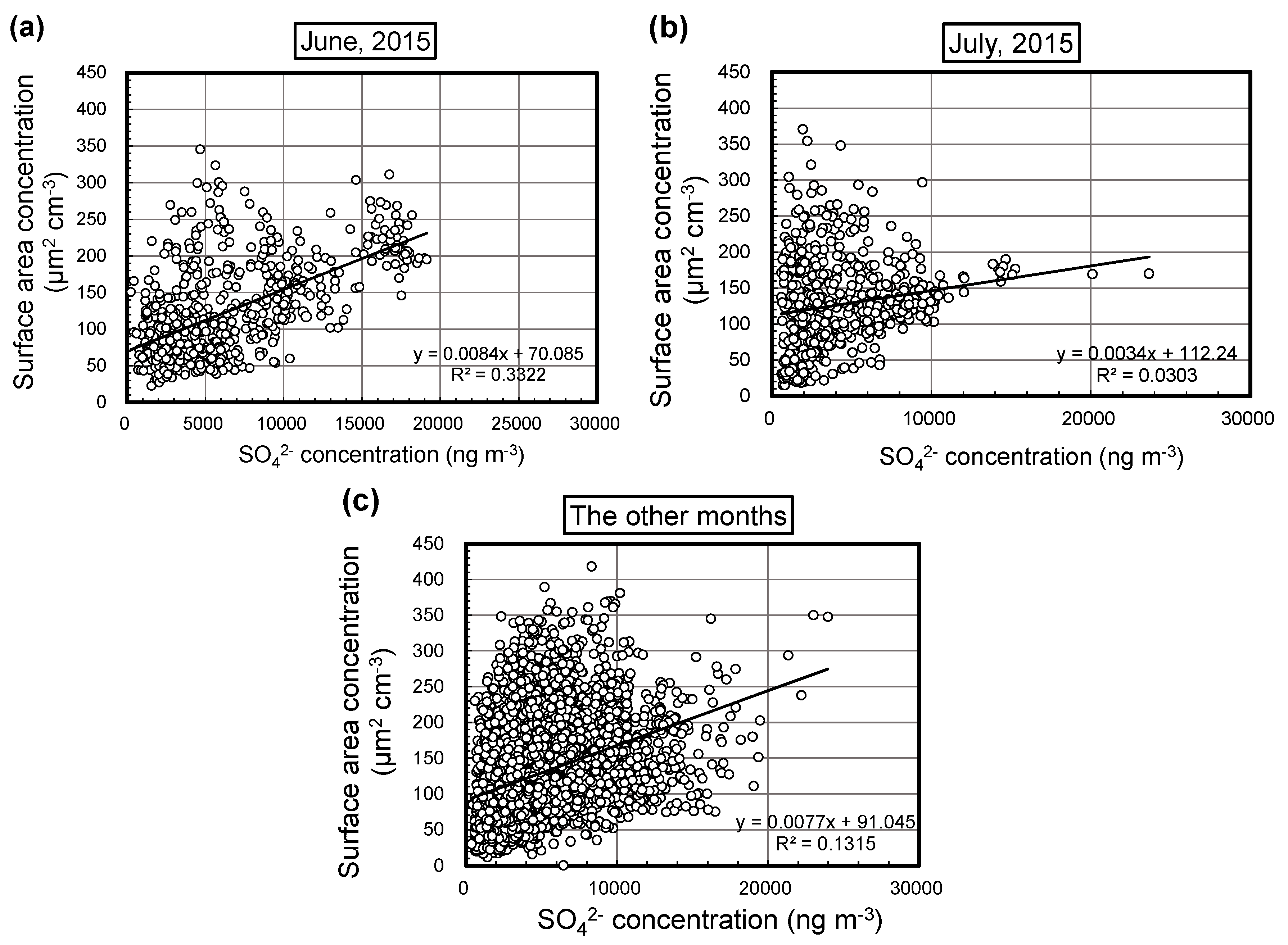
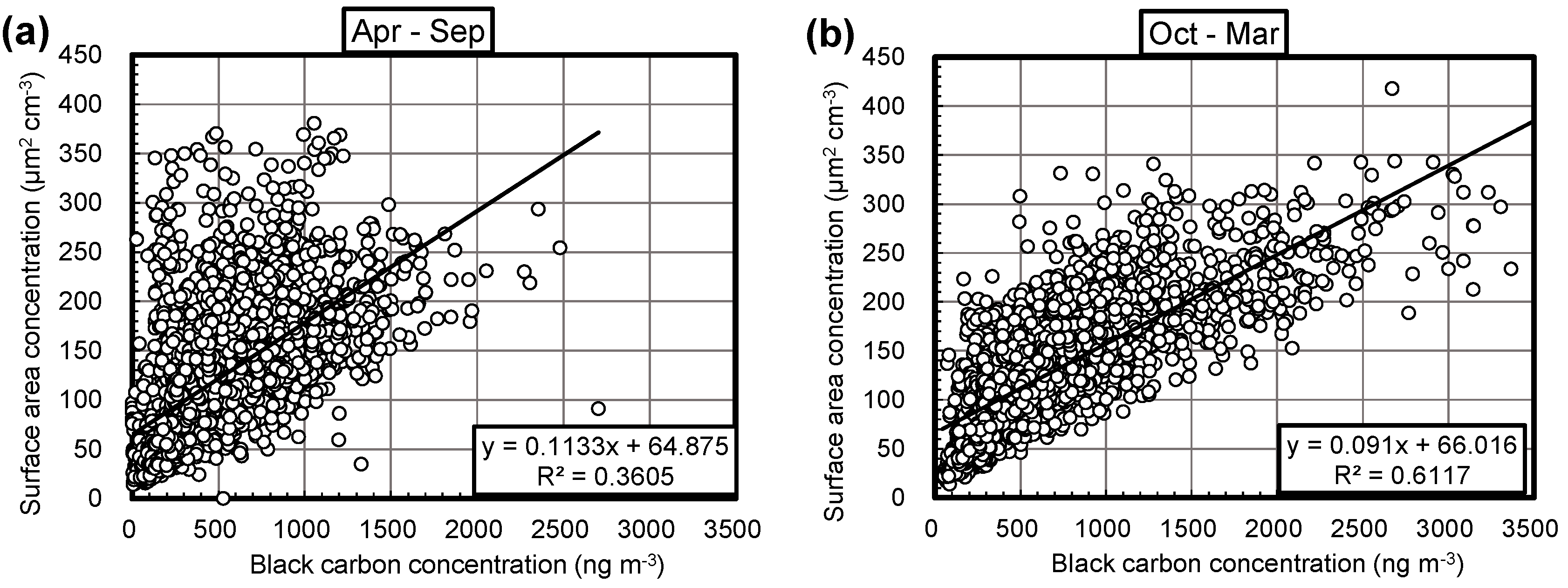
3.2. Monthly Correlations between Aerosol Surface Area and BC Concentrations
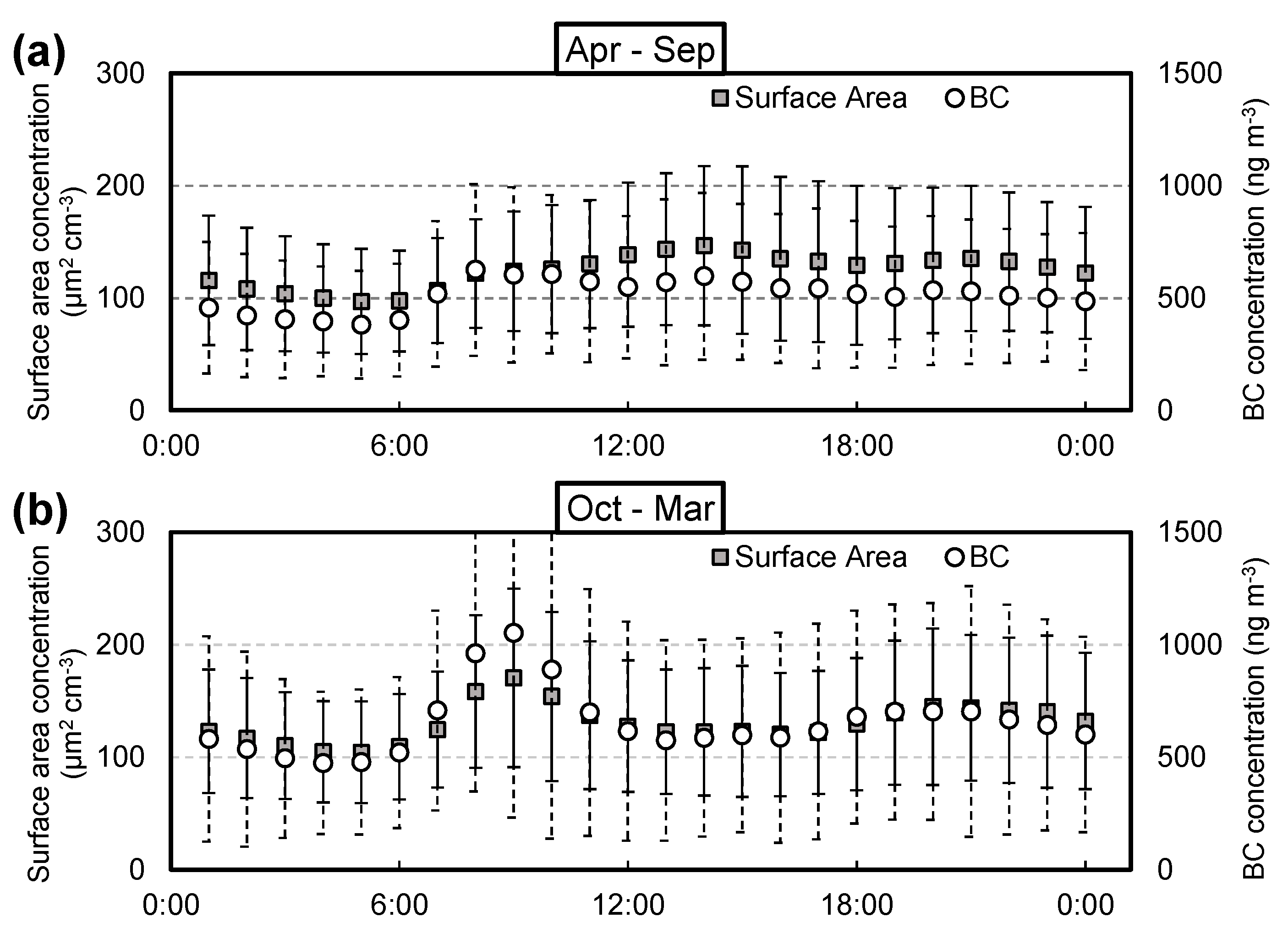
3.3. Effect of Land and Sea Breeze on the Correlation between Aerosol Surface Area and Black Carbon Concentrations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dockery, D.W.; Pope, C.A., III; Xu, X.; Spengler, J.D.; Ware, J.H.; Fay, M.E.; Ferris, B.G., Jr.; Speizer, F.E. An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 1993, 329, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A., III; Thun, M.J.; Namboodiri, M.M.; Dockery, D.W.; Evans, J.S.; Speizer, F.E.; Heath, C.W., Jr. Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am. J. Respir. Crit. Care Med. 1995, 151, 669–674. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Air Pollution and Cancer; IARC Scientific Publication: Lyon, France, 2013; No. 161. [Google Scholar]
- Ferin, J.; Oberdörster, G.; Penney, D.P. Pulmonary retention of ultrafine and fine particles in rats. Am. J. Respir. Cell Mol. Biol. 1992, 6, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Oberdörster, G.; Gelein, R.M.; Ferin, J.; Weiss, B. Association of particulate air pollution and acute mortality: Involvement of ultrafine particles? Inhal. Toxicol. 1995, 7, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, K.; Li, X.Y.; MacNee, W. Ultrafine (nanometre) particle mediated lung injury. J. Aerosol Sci. 1998, 29, 553–560. [Google Scholar] [CrossRef]
- The National Institute for Occupational Safety and Health (NIOSH). The National Institute for Occupational Safety and Health (NIOSH): Approaches to Safe Nanotechnology; DHHS (NIOSH) Publication No. 2009-125; The National Institute for Occupational Safety and Health (NIOSH): Washington, DC, USA, 2009. [Google Scholar]
- Oberdörster, G.; Finkelstein, J.N.; Johnston, C.; Gelein, R.; Cox, C.; Baggs, R.; Elder, A.C.P. Acute pulmonary effects of ultrafine particles in rats and mice. Res. Rep. Health Effects Inst. 2000, 96, 5–74. [Google Scholar]
- Oberdörster, G.; Oberdörster, E.; Oberdörster, J. Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005, 113, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, J. Risk Assessment of Manufactured Nanomaterials: Carbon Nanotubes (CNT); Final Report Issued on 12 August 2011, Executive Summary; NEDO Project “Research and Development of Nanoparticle Characterization Methods.” (P06041); New Energy and Industrial Technology Development Organization: Kawasaki, Japan, 2011. [Google Scholar]
- Schmid, O.; Stoeger, T. Surface area is the biologically most effective dose metric for acute nanoparticle toxicity in the lung. J. Aerosol Sci. 2016, 99, 133–143. [Google Scholar] [CrossRef]
- Oberdörster, G. Pulmonary effects of inhaled ultrafine particles. Int. Arch. Occup. Environ. Health 2001, 74, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giechaskiel, B.; Alföldy, B.; Drossinos, Y. A metric for health effects studies of diesel exhaust particles. J. Aerosol Sci. 2009, 40, 639–651. [Google Scholar] [CrossRef]
- Skuland, T.; Øvrevik, J.; Låg, M.; Refsnes, M. Role of size and surface area for pro-inflammatory responses to silica nanoparticles in epithelial lung cells: Importance of exposure conditions. Toxicol. In Vitro 2014, 28, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Okuda, T. Measurement of the specific surface area and particle size distribution of atmospheric aerosol reference materials. Atmos. Environ. 2013, 75, 1–5. [Google Scholar] [CrossRef]
- Okuda, T.; Isobe, R.; Nagai, Y.; Okahisa, S.; Funato, K.; Inoue, K. Development of a high-volume PM2.5 particle sampler using impactor and cyclone techniques. Aerosol Air Qual. Res. 2015, 15, 759–767. [Google Scholar] [CrossRef]
- Hatoya, K.; Okuda, T.; Funato, K.; Inoue, K. On-line measurement of the surface area concentration of aerosols in Yokohama, Japan, using the diffusion charging method. Asian J. Atmos. Environ. 2016, 10, 1–12. [Google Scholar] [CrossRef]
- Jung, H.; Kittelson, D.B. Characterization of aerosol surface instruments in transition regime. Aerosol Sci. Technol. 2005, 39, 902–911. [Google Scholar] [CrossRef]
- Fissan, H.; Neumann, S.; Trampe, A.; Pui, D.Y.H.; Shin, W.G. Rationale and principle of an instrument measuring lung deposited nanoparticle surface area. J. Nanopart. Res. 2007, 9, 53–59. [Google Scholar] [CrossRef]
- Shin, W.G.; Pui, D.Y.H.; Fissan, H.; Neumann, S.; Trampe, A. Calibration and numerical simulation of Nanoparticle Surface Area Monitor (TSI Model 3550 NSAM). J. Nanopart. Res. 2007, 9, 61–69. [Google Scholar] [CrossRef]
- Heitbrink, W.A.; Evans, D.E.; Ku, B.K.; Maynard, A.D.; Slavin, T.J.; Peters, T.M. Relationships among particle number, surface area, and respirable mass concentrations in automotive engine manufacturing. J. Occup. Environ. Hyg. 2009, 6, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yamazaki, H.; Hatoya, K.; Kaneyasu, N.; Yoshino, A.; Takami, A.; Funato, K.; Inoue, K.; Nishita, C.; Hara, K.; et al. Factors Controlling the Variation of Aerosol Surface Area Concentrations Measured by a Diffusion Charger in Fukuoka, Japan. Atmosphere 2016, 7, 33. [Google Scholar] [CrossRef]
- Velasco, E.; Siegmann, P.; Siegmann, H.C. Exploratory study of particle-bound polycyclic aromatic hydrocarbons in different environments of Mexico City. Atmos. Environ. 2004, 38, 4957–4968. [Google Scholar] [CrossRef]
- Ntziachristos, L.; Polidori, A.; Phuleria, H.; Geller, M.D.; Sioutas, C. Application of a diffusion charger for the measurement of particle surface concentration in different environments. Aerosol Sci. Technol. 2007, 41, 571–580. [Google Scholar] [CrossRef]
- Asbach, C.; Fissan, H.; Stahlmecke, B.; Kuhlbusch, T.A.J.; Pui, D.Y.H. Conceptual limitations and extensions of lung-deposited Nanoparticle Surface Area Monitor (NSAM). J. Nanopart. Res. 2009, 11, 101–109. [Google Scholar] [CrossRef]
- Albuquerque, P.C.; Gomes, J.F.; Bordado, J.C. Assessment of exposure to airborne ultrafine particles in the urban environment of Lisbon, Portugal. J. Air Waste Manag. Assoc. 2012, 62, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.F.P.; Albuquerque, P.C.S.; Esteves, H.M.D.S.; Carvalho, P.A. Notice on a methodology for characterizing emissions of ultrafine particles/nanoparticles in microenvironments. Energy Emiss. Cont. Technol. 2013, 1, 15–27. [Google Scholar] [CrossRef]
- China, S.; Salvadori, N.; Mazzoleni, C. Effect of traffic and driving characteristics on morphology of atmospheric soot particles at freeway on-ramps. Environ. Sci. Technol. 2014, 48, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Kaneyasu, N.; Yamamoto, S.; Sato, K.; Takami, A.; Hayashi, M.; Hara, K.; Kawamoto, K.; Okuda, T.; Hatakeyama, S. Impact of long-range transport of aerosols on the PM2.5 composition at a major metropolitan area in the northern Kyushu area of Japan. Atmos. Environ. 2014, 97, 416–425. [Google Scholar] [CrossRef]
- Takami, A.; Miyoshi, T.; Irei, S.; Yoshino, A.; Sato, K.; Shimizu, A.; Hayashi, M.; Hara, K.; Kaneyasu, N.; Hatakeyama, S. Analysis of organic aerosol in Fukuoka, Japan using a PMF method. Aerosol Air Qual. Res. 2016, 16, 314–322. [Google Scholar] [CrossRef]
- Yoshino, A.; Takami, A.; Sato, K.; Shimizu, A.; Kaneyasu, N.; Hatakeyama, S.; Hara, K.; Hayashi, M. Influence of Trans-Boundary Air Pollution on the Urban Atmosphere in Fukuoka, Japan. Atmosphere 2016, 7, 51. [Google Scholar] [CrossRef]
- Kaneyasu, N.; Takami, A.; Sato, K.; Hatakeyama, S.; Hara, S.; Kawamoto, K.; Yamamoto, S. Year-round behavior of PM2.5 in a remote island and urban site in the northern Kyushu area, Japan. J. Jpn. Soc. Atmos. Environ. 2011, 46, 111–118. (In Japanese) [Google Scholar]
- International Commission on Radiological Protection (ICRP). Human respiratory tract model for radiological protection. Ann. ICRP 1994, 24, 1–482. [Google Scholar]
- Venkatachari, P.; Zhou, L.; Hopke, P.K.; Schwab, J.J.; Demerjian, K.L.; Weimer, S.; Hogrefe, O.; Felton, D.; Rattigan, O. An intercomparison of measurement methods for carbonaceous aerosol in the ambient air in New York City. Aerosol Sci. Technol. 2006, 40, 788–795. [Google Scholar] [CrossRef]
- Ng, I.P.; Ma, H.; Kittelson, D.B.; Miller, A.L. Comparing Measurements of Carbon in Diesel Exhaust Aerosols Using the Aethalometer, NIOSH Method 5040, and SMPS; SAE Technical Paper Series 2007-01-0334; University of Minnesota: Minneapolis, MN, USA, 2007. [Google Scholar]
- Osada, K.; Kamiguchi, Y.; Yamamoto, S.; Kuwahara, S.; Pan, X.; Hara, Y.; Uno, I. Comparison of ionic concentrations on size-segregated atmospheric aerosol particles based on a denuder-filter method and a Continuous Dichotomous Aerosol Chemical Speciation Analyzer (ACSA-12). Earozoru Kenkyu 2016, 31, 203–209. (In Japanese) [Google Scholar]
- Fukuoka Prefecture Website. Available online: http://www.fihes.pref.fukuoka.jp/taiki-new/Nipo/OyWbNpKm0151.htm (accessed on 6 May 2017).
- Japan Meteorological Agency Website. Available online: http://www.data.jma.go.jp/obd/stats/etrn/index.php (accessed on 6 May 2017).
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT atmospheric transport and dispersion modeling system. Bull. Am. Meteor. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.D. Real-Time Environmental Applications and Display sYstem (READY) Website. NOAA Air Resources Laboratory: Silver Spring, MD, USA, 2017. Available online: http://ready.arl.noaa.gov/HYSPLIT.php (accessed on 26 June 2017).
- China, S.; Scarnato, B.; Owen, R.C.; Zhang, B.; Ampadu, M.T.; Kumar, S.; Dzepina, K.; Dziobak, M.P.; Fialho, P.; Perlinger, J.A.; et al. Morphology and mixing state of aged soot particles at a remote marine free troposphere site: Implications for optical properties. Geophys. Res. Lett. 2015, 42, 1243–1250. [Google Scholar] [CrossRef]
- Shiraiwa, M.; Kondo, Y.; Moteki, N.; Takegawa, N.; Sahu, L.K.; Takami, A.; Hatakeyama, S.; Yonemura, S.; Blake, D.R. Radiative impact of mixing state of black carbon aerosol in Asian outflow. J. Geophys. Res. 2008, 113, D24210. [Google Scholar] [CrossRef]
- Takami, A.; Mayama, N.; Sakamoto, T.; Ohishi, K.; Irei, S.; Yoshino, A.; Hatakeyama, S.; Murano, K.; Sadanaga, Y.; Bandow, H.; et al. Structural analysis of aerosol particles by microscopic observation using a time-of-flight secondary ion mass spectrometer. J. Geophys. Res. Atmos. 2013, 118, 6726–6737. [Google Scholar] [CrossRef]
- Moffet, R.C.; O’Brien, R.E.; Alpert, P.A.; Kelly, S.T.; Pham, D.Q.; Gilles, M.K.; Knopf, D.A.; Laskin, A. Morphology and mixing of black carbon particles collected in central California during the CARES field study. Atmos. Chem. Phys. 2016, 16, 14515–14525. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Unit | Mean | SD | n |
|---|---|---|---|---|
| Surface Area | µm2·cm−3 | 127 | 62 | 8032 |
| PM2.5 mass | µg·m−3 | 17.3 | 10.7 | 8064 |
| BC | ng·m−3 | 579 | 430 | 7698 |
| SO42− | µg·m−3 | 4.92 | 3.22 | 7511 |
| Particle Number (>0.3 μm) | # cm−3 | 419 | 469 | 7281 |
| Surface Area vs. BC | |||||
| Year | Month | Slope (µm2·cm−3)/(ng·m−3) | Intercept µm2·cm−3 | Coefficient of Determination | n |
| 2015 | April | 0.12 | 40 | 0.45 | 489 |
| May | 0.093 | 81 | 0.41 | 659 | |
| June | 0.11 | 73 | 0.23 | 696 | |
| July | 0.10 | 100 | 0.23 | 625 | |
| August | 0.15 | 44 | 0.42 | 511 | |
| September | 0.17 | 39 | 0.62 | 678 | |
| October | 0.088 | 77 | 0.54 | 623 | |
| November | 0.10 | 51 | 0.59 | 546 | |
| December | 0.089 | 55 | 0.76 | 374 | |
| 2016 | January | 0.079 | 64 | 0.71 | 535 |
| February | 0.12 | 57 | 0.66 | 713 | |
| March | 0.13 | 46 | 0.64 | 632 | |
| Surface Area vs. SO42− | |||||
| Year | Month | Slope (µm2·cm−3)/(ng·m−3) | Intercept µm2·cm−3 | Coefficient of Determination | n |
| 2015 | April | 0.0072 | 72 | 0.21 | 508 |
| May | 0.0068 | 99 | 0.10 | 651 | |
| June | 0.0084 | 70 | 0.33 | 676 | |
| July | 0.0034 | 112 | 0.030 | 570 | |
| August | 0.0056 | 87 | 0.15 | 461 | |
| September | 0.014 | 65 | 0.22 | 641 | |
| October | 0.0073 | 110 | 0.15 | 642 | |
| November | 0.017 | 79 | 0.12 | 428 | |
| December | 0.013 | 78 | 0.18 | 343 | |
| 2016 | January | 0.012 | 68 | 0.23 | 679 |
| February | 0.0066 | 100 | 0.084 | 655 | |
| March | 0.0084 | 80 | 0.19 | 683 | |
| (a) Night (0:00–6:00) | (b) Morning (6:00–12:00) | ||||||||
| Year | Month | Slope (µm2·cm−3)/(ng·m−3) | Intercept µm2·cm−3 | Coefficient of Determination | n | Slope (µm2·cm−3)/(ng·m−3) | Intercept µm2·cm−3 | Coefficient of Determination | n |
| 2015 | Apr | 0.12 | 30 | 0.87 | 119 | 0.068 | 70 | 0.17 | 112 |
| May | 0.12 | 62 | 0.47 | 172 | 0.070 | 83 | 0.42 | 160 | |
| Jun | 0.17 | 52 | 0.41 | 174 | 0.095 | 72 | 0.27 | 173 | |
| Jul | 0.15 | 36 | 0.63 | 163 | 0.051 | 102 | 0.09 | 149 | |
| Aug | 0.20 | 24 | 0.70 | 136 | 0.094 | 68 | 0.25 | 124 | |
| Sep | 0.19 | 28 | 0.70 | 164 | 0.12 | 55 | 0.48 | 160 | |
| Oct | 0.10 | 69 | 0.60 | 161 | 0.085 | 74 | 0.70 | 153 | |
| Nov | 0.13 | 40 | 0.63 | 138 | 0.088 | 51 | 0.74 | 135 | |
| Dec | 0.091 | 45 | 0.78 | 92 | 0.085 | 47 | 0.84 | 89 | |
| 2016 | Jan | 0.080 | 51 | 0.68 | 132 | 0.083 | 65 | 0.76 | 132 |
| Feb | 0.11 | 59 | 0.64 | 174 | 0.12 | 60 | 0.74 | 169 | |
| Mar | 0.13 | 47 | 0.50 | 156 | 0.11 | 57 | 0.61 | 156 | |
| (c) Afternoon (12:00–18:00) | (d) Evening (18:00–24:00) | ||||||||
| Year | Month | Slope (µm2·cm−3)/(ng·m−3) | Intercept µm2·cm−3 | Coefficient of Determination | n | Slope (µm2·cm−3)/(ng·m−3) | Intercept µm2·cm−3 | Coefficient of Determination | n |
| 2015 | Apr | 0.12 | 50 | 0.31 | 128 | 0.16 | 21 | 0.68 | 130 |
| May | 0.084 | 95 | 0.30 | 160 | 0.12 | 74 | 0.62 | 167 | |
| Jun | 0.072 | 99 | 0.13 | 178 | 0.15 | 78 | 0.29 | 171 | |
| Jul | 0.060 | 119 | 0.07 | 155 | 0.14 | 53 | 0.45 | 157 | |
| Aug | 0.15 | 48 | 0.34 | 122 | 0.22 | 13 | 0.59 | 123 | |
| Sep | 0.18 | 34 | 0.62 | 174 | 0.21 | 27 | 0.71 | 168 | |
| Oct | 0.087 | 79 | 0.32 | 151 | 0.091 | 82 | 0.53 | 158 | |
| Nov | 0.096 | 61 | 0.36 | 140 | 0.13 | 43 | 0.72 | 133 | |
| Dec | 0.089 | 56 | 0.71 | 95 | 0.10 | 64 | 0.81 | 98 | |
| 2016 | Jan | 0.063 | 73 | 0.63 | 133 | 0.083 | 73 | 0.71 | 138 |
| Feb | 0.099 | 61 | 0.49 | 172 | 0.15 | 49 | 0.69 | 174 | |
| Mar | 0.12 | 46 | 0.56 | 159 | 0.17 | 29 | 0.81 | 161 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiriya, M.; Okuda, T.; Yamazaki, H.; Hatoya, K.; Kaneyasu, N.; Uno, I.; Nishita, C.; Hara, K.; Hayashi, M.; Funato, K.; et al. Monthly and Diurnal Variation of the Concentrations of Aerosol Surface Area in Fukuoka, Japan, Measured by Diffusion Charging Method. Atmosphere 2017, 8, 114. https://doi.org/10.3390/atmos8070114
Kiriya M, Okuda T, Yamazaki H, Hatoya K, Kaneyasu N, Uno I, Nishita C, Hara K, Hayashi M, Funato K, et al. Monthly and Diurnal Variation of the Concentrations of Aerosol Surface Area in Fukuoka, Japan, Measured by Diffusion Charging Method. Atmosphere. 2017; 8(7):114. https://doi.org/10.3390/atmos8070114
Chicago/Turabian StyleKiriya, Miho, Tomoaki Okuda, Hana Yamazaki, Kazuki Hatoya, Naoki Kaneyasu, Itsushi Uno, Chiharu Nishita, Keiichiro Hara, Masahiko Hayashi, Koji Funato, and et al. 2017. "Monthly and Diurnal Variation of the Concentrations of Aerosol Surface Area in Fukuoka, Japan, Measured by Diffusion Charging Method" Atmosphere 8, no. 7: 114. https://doi.org/10.3390/atmos8070114
APA StyleKiriya, M., Okuda, T., Yamazaki, H., Hatoya, K., Kaneyasu, N., Uno, I., Nishita, C., Hara, K., Hayashi, M., Funato, K., Inoue, K., Yamamoto, S., Yoshino, A., & Takami, A. (2017). Monthly and Diurnal Variation of the Concentrations of Aerosol Surface Area in Fukuoka, Japan, Measured by Diffusion Charging Method. Atmosphere, 8(7), 114. https://doi.org/10.3390/atmos8070114