
Examining the Impact of Dimethyl Sulfide Emissions on Atmospheric Sulfate over the Continental U.S.


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Base Model Configuration
2.2. DMS Emission Implementation
2.3. DMS Chemistry Implementation
2.4. Boundary Conditions
3. Results and Discussion
3.1. Annual Impact of DMS Emissions on Surface Inorganic Particles
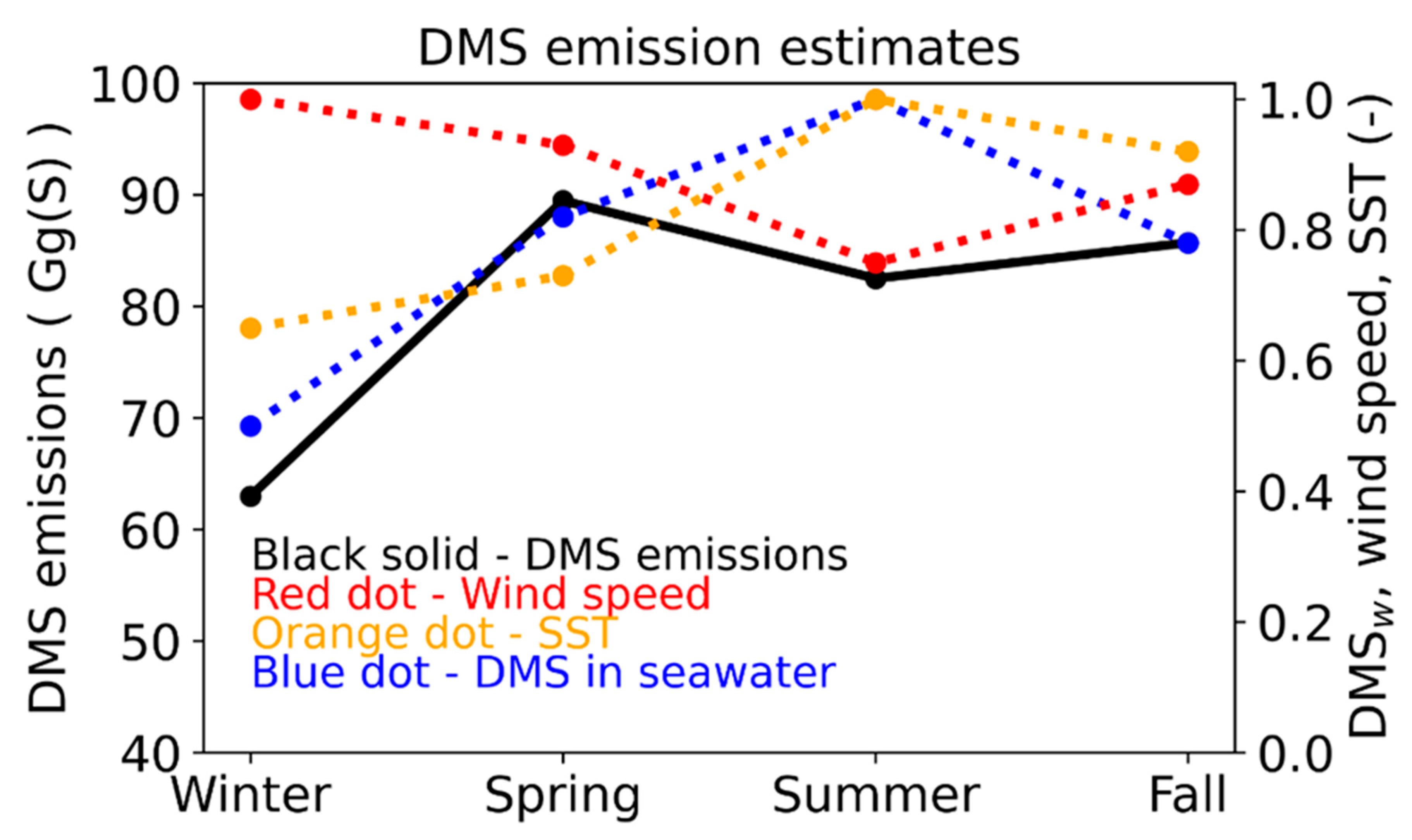
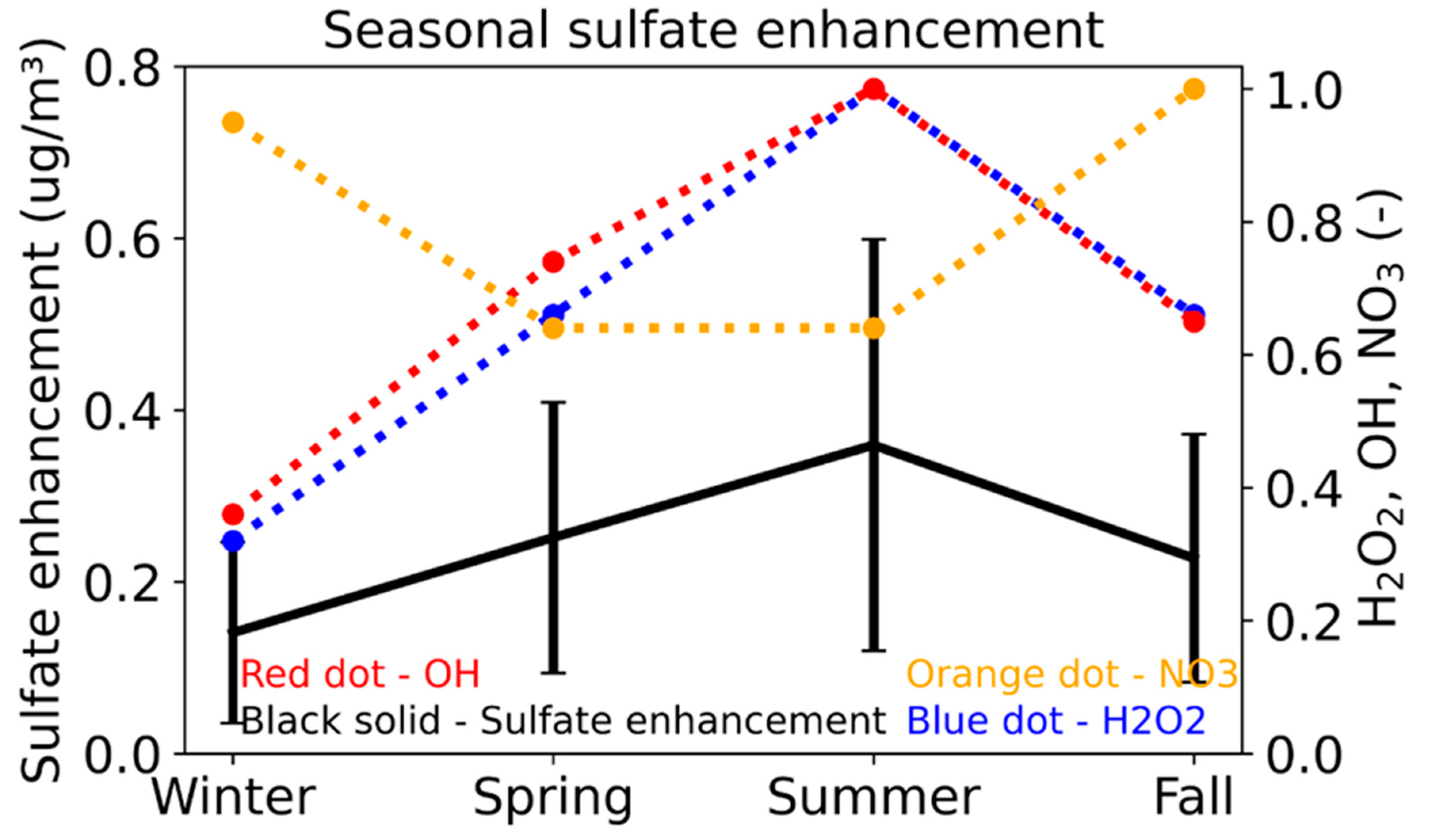
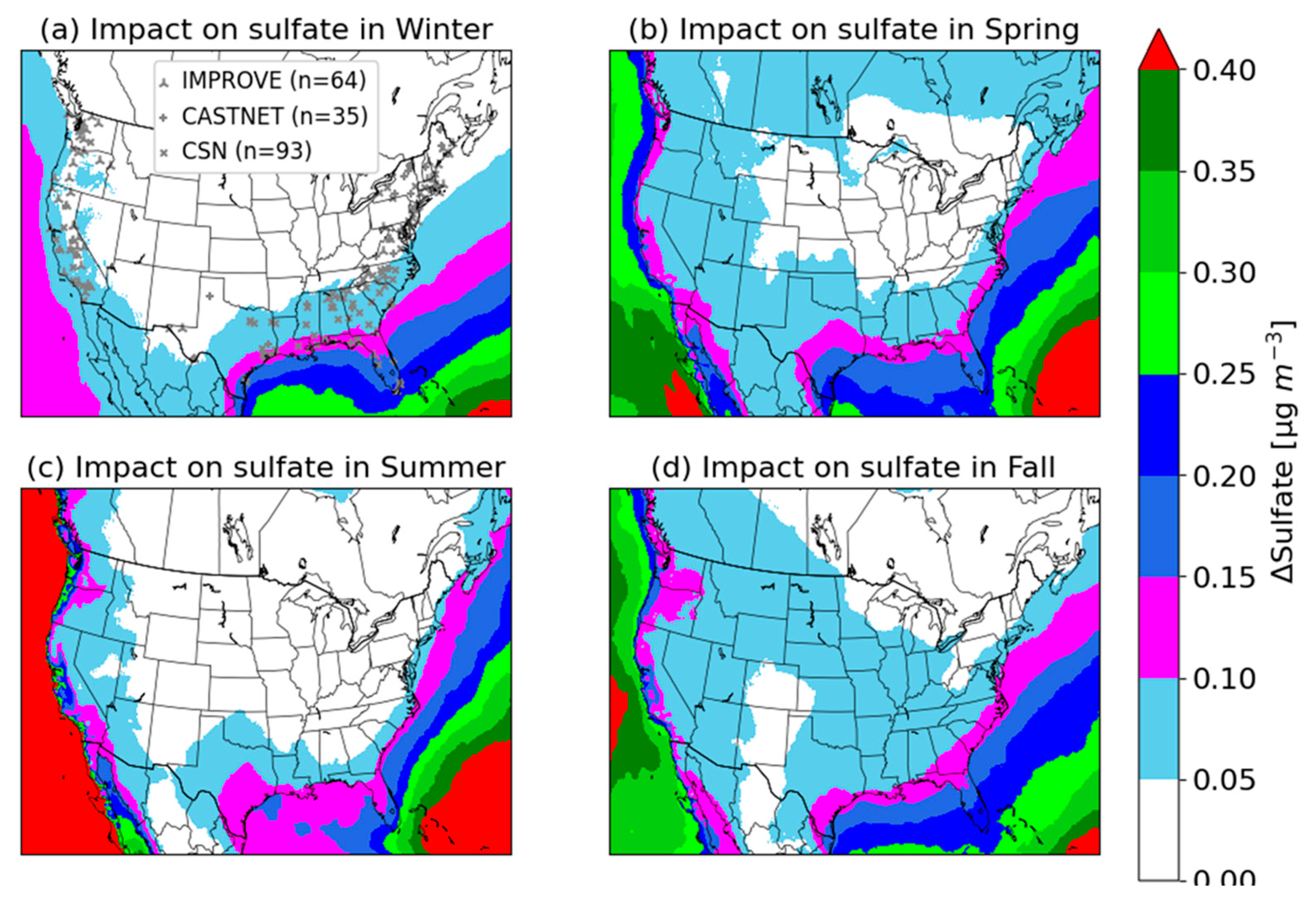
3.2. Seasonal Impacts of DMS Emissions on Surface Sulfate
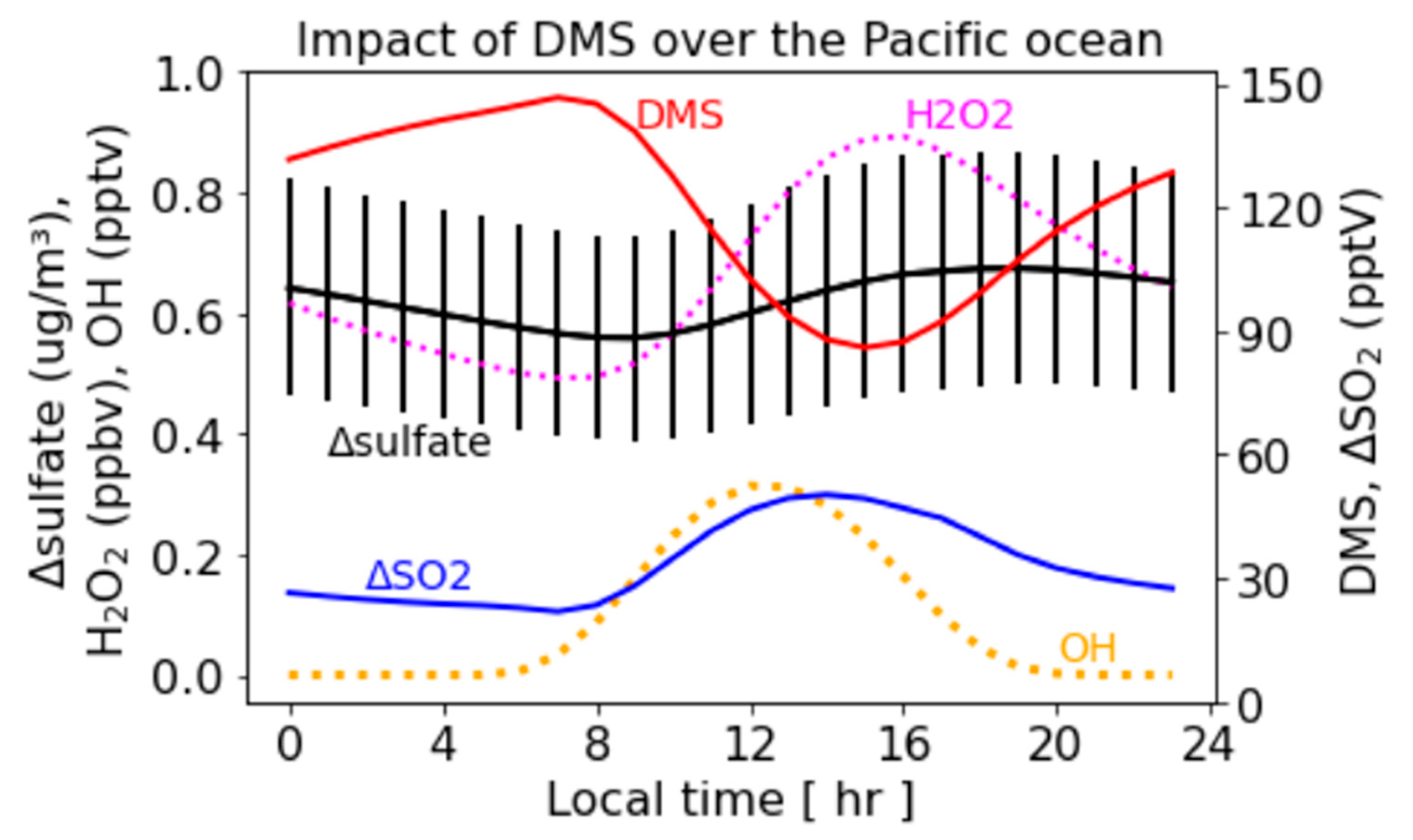
3.3. Diurnal Variation in the Sulfate Enhancement
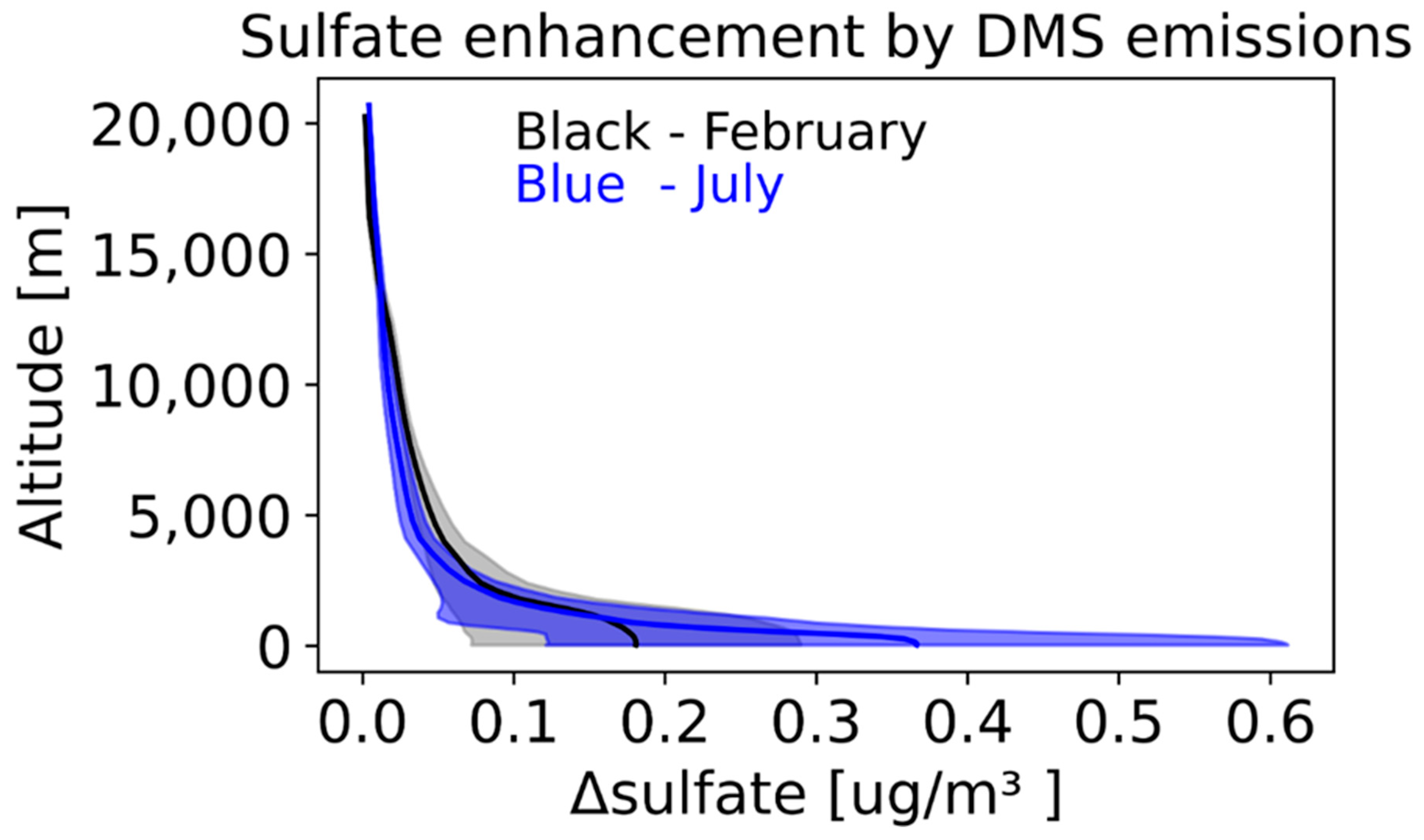
3.4. Impact of DMS Emissions on Sulfate Aloft
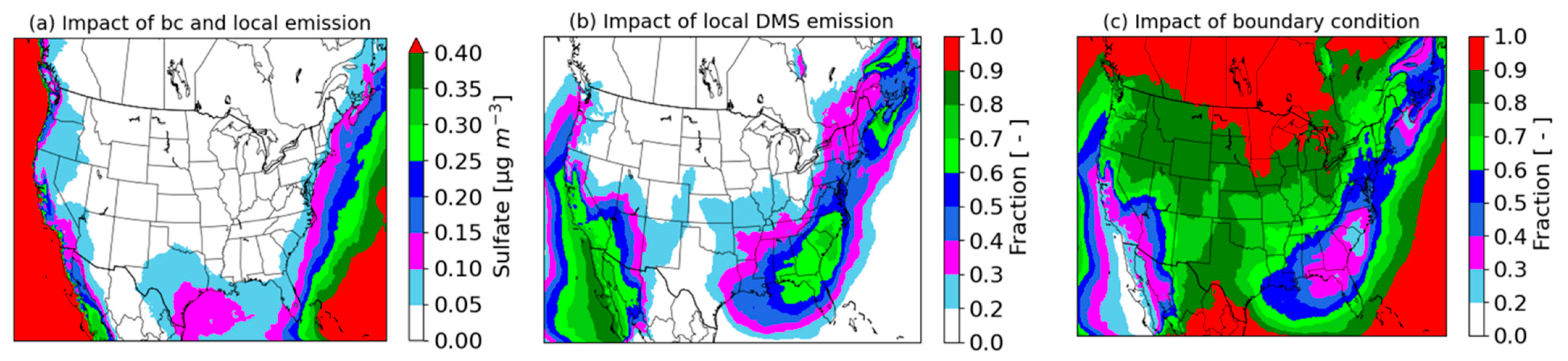
3.5. Impact of DMS Emissions and Boundary Conditions on Sulfate
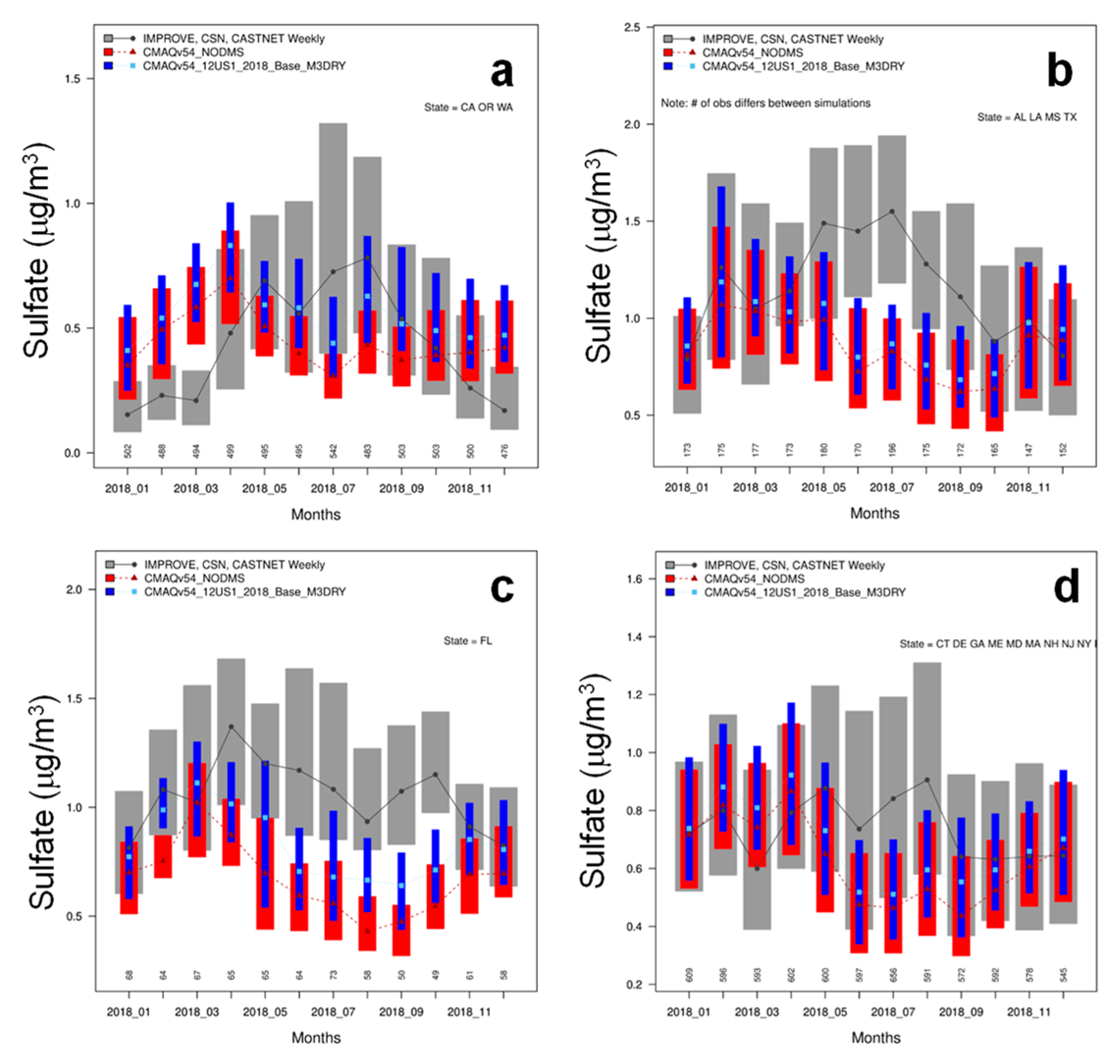
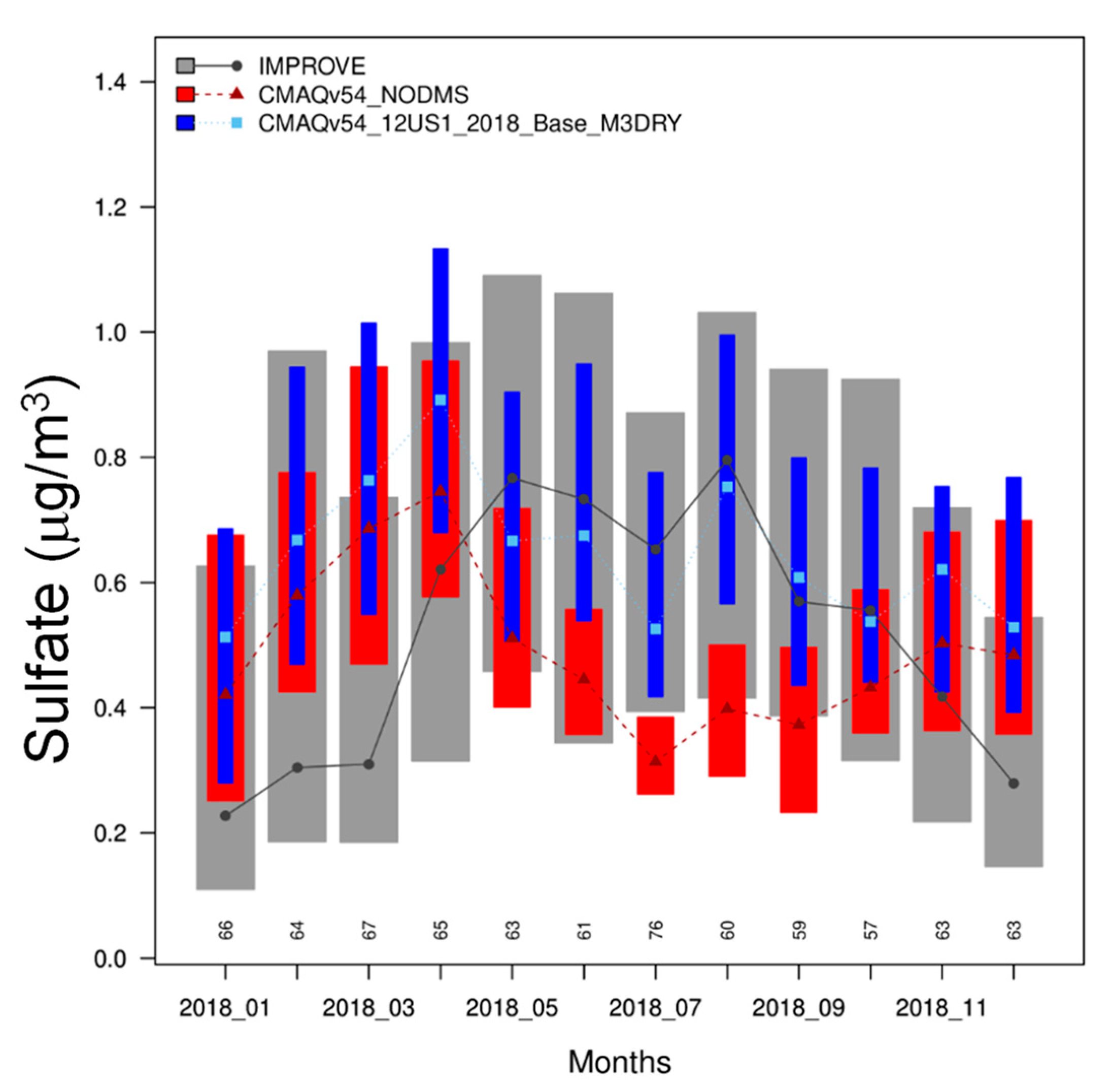
3.6. Comparison with Observed Sulfate
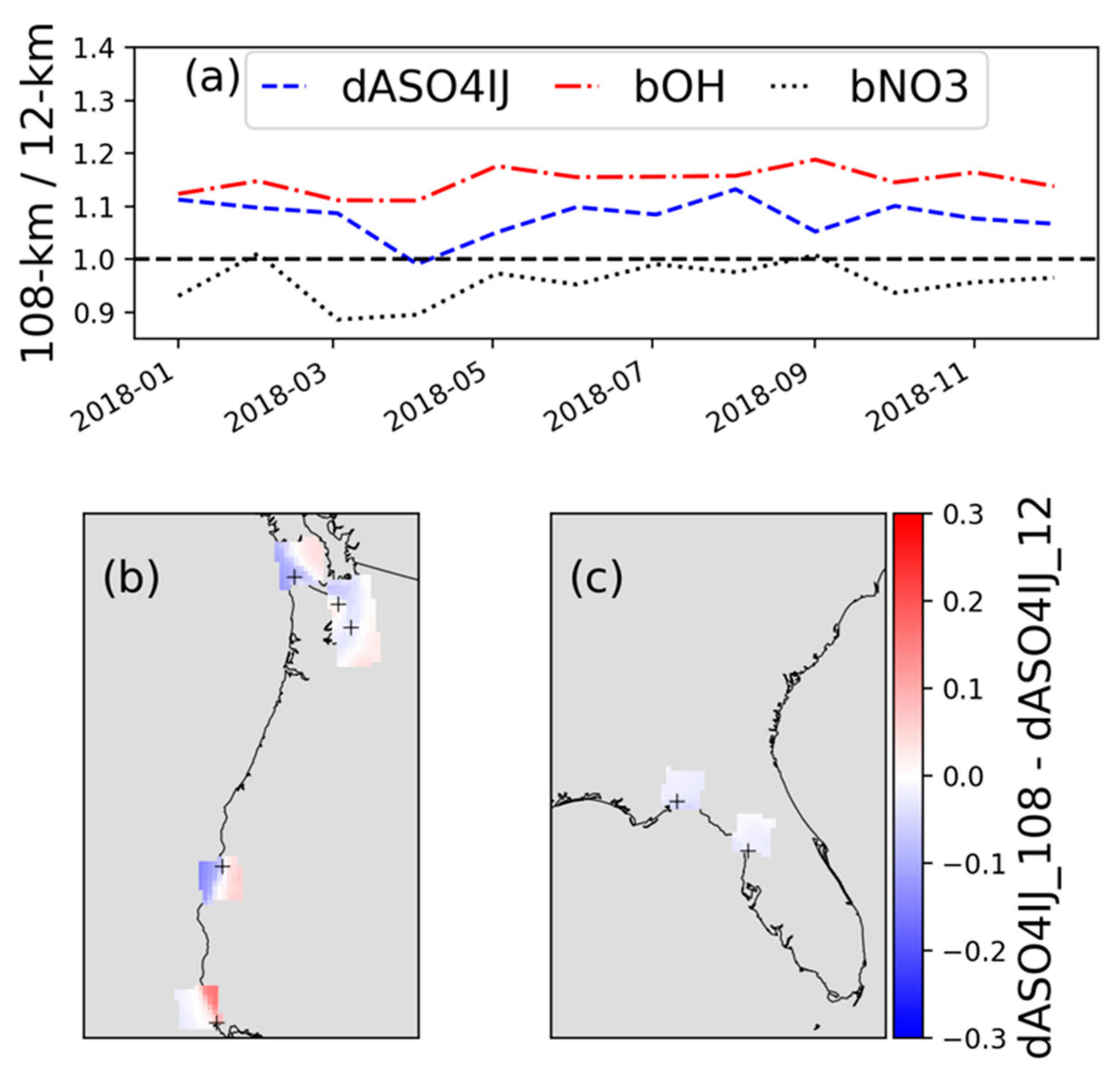
3.7. Changes in DMS-Initiated Sulfate Enhancement with Horizontal Grid Resolution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carpenter, L.J.; Archer, S.D.; Beale, R. Ocean-atmosphere trace gas exchange. Chem. Soc. Rev. 2012, 41, 6473–6506. [Google Scholar] [CrossRef] [PubMed]
- Charlson, R.J.; Lovelock, J.E.; Andreae, M.O.; Warren, S.G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 1987, 326, 655–661. [Google Scholar] [CrossRef]
- Lana, A.; Bell, T.G.; Simó, R.; Vallina, S.M.; Ballabrera-Poy, J.; Kettle, A.J.; Dachs, J.; Bopp, L.; Saltzman, E.S.; Stefels, J.; et al. An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Glob. Biogeochem. Cycles 2011, 25. [Google Scholar] [CrossRef]
- Fung, K.M.; Heald, C.L.; Kroll, J.H.; Wang, S.; Jo, D.S.; Gettelman, A.; Lu, Z.; Liu, X.; Zaveri, R.A.; Apel, E.C.; et al. Exploring dimethyl sulfide (DMS) oxidation and implications for global aerosol radiative forcing. Atmos. Chem. Phys. 2022, 22, 1549–1573. [Google Scholar] [CrossRef]
- Aas, W.; Mortier, A.; Bowersox, V.; Cherian, R.; Faluvegi, G.; Fagerli, H.; Hand, J.; Klimont, Z.; Galy-Lacaux, C.; Lehmann, C.M.B.; et al. Global and regional trends of atmospheric sulfur. Nat. Sci. Rep. 2019, 9, 953. [Google Scholar] [CrossRef]
- Tanner, R.L.; Bairai, S.T.; Mueller, S.F. Trends in concentrations of atmospheric gaseous and particulate species in rural eastern Tennessee as related to primary emission reductions. Atmos. Chem. Phys. 2015, 15, 9781–9797. [Google Scholar] [CrossRef]
- Malm, W.C.; Schichtel, B.A.; Hand, J.L.; Collett, J.L., Jr. Concurrent temporal and spatial trends in sulfate and organic mass concentrations measured in the IMPROVE Monitoring Program. J. Geophys. Res. Atmos. 2017, 122, 10462–10476. [Google Scholar] [CrossRef]
- Avise, J.; Chen, J.; Turkiewicz, K.; DaMassa, J.; Vanderspek, S. Wintertime PM2.5 Pollution in California, EM, The Magazine for Environmental Managers, A&WMA, December 2019. Available online: www.awma.org/em/ (accessed on 23 January 2023).
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From air pollution to Climate Change; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Lee, C.L.; Brimblecombe, P. Anthropogenic contributions to global carbonyl sulfide, carbon disulfide and organosulfides fluxes. Earth-Sci. Rev. 2016, 160, 1–18. [Google Scholar] [CrossRef]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.-M.; Kondo, Y.; Liao, H.; Lohmann, U. Clouds and Aerosols, Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013; pp. 571–657. [Google Scholar] [CrossRef]
- Breider, T.J.; Chipperfield, M.P.; Richards, N.A.D.; Carslaw, K.S.; Mann, G.W.; Spracklen, D.V. Impact of BrO on dimethylsulfide in the remote marine boundary layer. Geophys. Res. Lett. 2010, 37, L02807. [Google Scholar] [CrossRef]
- Park, R.J.; Jacob, D.J.; Field, B.D.; Yantosca, R.M.; Chin, M. Natural and transboundary pollution influences on sulfate-nitrate-ammonium aerosols in the United States: Implications for policy. J. Geophys. Res.-Atmos. 2004, 109, D15204. [Google Scholar] [CrossRef]
- Kloster, S.; Feichter, J.; Maier-Reimer, E.; Six, K.D.; Stier, P.; Wetzel, P. DMS cycle in the marine ocean-atmosphere system; a global model study. Biogeosciences 2006, 3, 29–51. [Google Scholar] [CrossRef]
- Thomas, M.A.; Suntharalingam, P.; Pozzoli, L.; Rast, S.; Devasthale, A.; Kloster, S.; Feichter, J.; Lenton, T.M. Quantification of DMS aerosol-cloud-climate interactions using the ECHAM5-HAMMOZ model in a current climate scenario. Atmos. Chem. Phys. 2010, 10, 7425–7438. [Google Scholar] [CrossRef]
- Chen, Q.; Sherwen, T.; Evans, M.; Alexander, B. DMS oxidation and sulfur aerosol formation in the marine troposphere: A focus on reactive halogen and multiphase chemistry. Atmos. Chem. Phys. 2018, 18, 13617–13637. [Google Scholar] [CrossRef]
- Itahashi, S.; Mathur, R.; Hogrefe, C.; Napelenok, S.L.; Zhang, Y. Incorporation of volcanic SO2 emissions in the Hemispheric CMAQ (H-CMAQ) version 5.2 modeling system and assessing their impacts on sulfate aerosol over the Northern Hemisphere. Geosci. Model Dev. 2021, 14, 5751–5768. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.F.; Mao, Q.; Mallard, J.W. Modeling natural emissions in the Community Multiscale Air Quality (CMAQ) model—Part 2: Modifications for simulating natural emissions. Atmos. Chem. Phys. 2011, 11, 293–320. [Google Scholar] [CrossRef]
- Perraud, V.; Horne, J.R.; Martinez, A.S.; Kalinowski, J.; Meinardi, S.; Dawson, M.L.; Wingen, L.M.; Dabdub, D.; Blake, D.R.; Gerber, R.B.; et al. The future of airborne sulfur-containing particles in the absence of fossil fuel sulfur dioxide emissions. Proc. Natl. Acad. Sci. USA 2015, 112, 13514–13519. [Google Scholar] [CrossRef]
- Zhao, J.; Sarwar, G.; Gantt, B.; Foley, K.; Henderson, B.H.; Pye, H.O.T.; Fahey, K.M.; Kang, D.; Mathur, R.; Zhang, Y.; et al. Impact of dimethylsulfide chemistry on air quality over the Northern Hemisphere. Atmos. Environ. 2021, 244, 117961. [Google Scholar] [CrossRef]
- Appel, K.W.; Bash, J.O.; Fahey, K.M.; Foley, K.M.; Gilliam, R.C.; Hogrefe, C.; Hutzell, W.T.; Kang, D.; Mathur, R.; Murphy, B.N.; et al. The Community Multiscale Air Quality (CMAQ) model versions 5.3 and 5.3.1: System updates and evaluation. Geosci. Model Dev. 2021, 14, 2867–2897. [Google Scholar] [CrossRef]
- Kitayama, K.; Morino, Y.; Yamaji, K.; Chatani, S. Uncertainties in O3 concentrations simulated by CMAQ over Japan using four chemical mechanisms. Atmos. Environ. 2019, 198, 448–462. [Google Scholar] [CrossRef]
- East, J.D.; Henderson, B.H.; Napelenok, S.L.; Koplitz, S.N.; Sarwar, G.; Gilliam, R.; Lenzen, A.; Tong, D.Q.; Pierce, R.B.; Garcia-Menendez, F. Inferring and evaluating satellite-based constraints on NOx emissions estimates in air quality simulations. Atmos. Chem. Phys. Discuss. 2022. in review. [Google Scholar] [CrossRef]
- Mathur, R.; Kang, D.; Napelenok, S.L.; Xing, J.; Hogrefe, C.; Sarwar, G.; Itahashi, S.; Henderson, B.H. How have divergent global emission trends influenced long-range transported ozone to North America? J. Geophys. Res. Atmos. 2022, 127, e2022JD036926. [Google Scholar] [CrossRef]
- Sarwar, G.; Christian Hogrefe, H.; Henderson, B.H.; Foley, K.; Mathur, R.; Murphy, B.; Ahmed, S. 2023 Characterizing variations in ambient PM25 concentrations at the, U.S. Embassy in Dhaka, Bangladesh using observations and the CMAQ modeling system. Atmos. Environ. 2023, 296, 119587. [Google Scholar] [CrossRef]
- US Environmental Protection Agency, 2022. CMAQ (Version 5.4) [Software]. Available online: https://zenodo.org/record/7218076#.ZCZVRHZBxPY (accessed on 25 March 2023).
- Skamarock, W.C.; Klemp, J.B. A time-split nonhydrostatic atmospheric model for weather research and forecasting applications. J. Comput. Phys. 2008, 227, 3465–3485. [Google Scholar] [CrossRef]
- Iacono, M.J.; Delamere, J.S.; Mlawer, E.J.; Shephard, M.W.; Clough, S.A.; Collins, W.D. Radiative forcing by long-lived greenhouse gases: Calculations with the AER radiative transfer models. J. Geophys. Res. Atmos. 2008, 113. [Google Scholar] [CrossRef]
- Kain, J.S. The Kain-Fritsch Convective Parameterization: An Update. J. Appl. Meteorol. 2004, 43, 170–181. [Google Scholar] [CrossRef]
- Heath, N.K.; Pleim, J.E.; Gilliam, R.C.; Kang, D. A simple lightning assimilation technique for improving retrospective WRF simulations. J. Adv. Model. Earth Syst. 2016, 8, 1806–1824. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Heath, N.K.; Gilliam, R.C.; Spero, T.L.; Pleim, J.E. Lightning assimilation in the WRF model (Version 4.1.1): Technique updates and assessment of the applications from regional to hemispheric scales. Geosci. Model Dev. 2022, 15, 8561–8579. [Google Scholar] [CrossRef]
- Morrison, H.; Gettelman, A. A new two-moment bulk stratiform cloud microphysics scheme in the Community Atmosphere Model, version 3 (CAM3). Part I: Description and numerical tests. J. Clim. 2008, 21, 3642–3659. [Google Scholar] [CrossRef]
- Pleim, J.E.; Xiu, A. Development and testing of a surface flux and planetary boundary layer model for application in mesoscale models. J. Appl. Meteorol. 1995, 34, 16–32. [Google Scholar] [CrossRef]
- Xiu, A.; Pleim, J.E. Development of a land surface model. Part I: Application in a mesoscale meteorological model. J. Appl. Meteorol. 2001, 40, 192–209. [Google Scholar] [CrossRef]
- Pleim, J.E. A combined local and nonlocal closure model for the atmospheric boundary layer. Part I: Model description and testing. J. Appl. Meteorol. Climatol. 2007, 46, 1383–1395. [Google Scholar] [CrossRef]
- Pleim, J.E. A combined local and nonlocal closure model for the atmospheric boundary layer. Part II: Application and evaluation in a mesoscale meteorological model. J. Appl. Meteorol. Climatol. 2007, 46, 1396–1409. [Google Scholar] [CrossRef]
- Gilliam, R.C.; Godowitch, J.M.; Rao, S.T. Improving the Horizontal Transport in the Lower Troposphere with Four Dimensional Data Assimilation (Vol. 53). Atmos. Environ. 2012, 53, 186–201. [Google Scholar] [CrossRef]
- Otte, T.L.; Pleim, J.E. The Meteorology-Chemistry Interface Processor (MCIP) for the CMAQ modeling system: Updates through MCIPv3.4.1. Geosci. Model Dev. 2010, 3, 243–256. [Google Scholar] [CrossRef]
- Torres-Vazquez, A.; Pleim, J.; Gilliam, R.; Pouliot, G. Performance evaluation of the meteorology and air quality conditions from multiscale WRF-CMAQ simulations for the Long Island Sound Tropospheric Ozone Study (LISTOS). J. Geophys. Res. Atmos. 2022, 127, e2021JD035890. [Google Scholar] [CrossRef] [PubMed]
- Yarwood, G.; Shi, Y.; Beardsley, R. Impact of CB6r5 Mechanism Changes on Air Pollutant Modeling in Texas. Final Report prepared for Texas Commission on Environmental Quality, Austin, TX 78753, USA; Ramboll US Corporation: Novato, CA, USA, 2020. [Google Scholar]
- Sarwar, G.; Simon, H.; Bhave, P.; Yarwood, G. Examining the impact of heterogeneous nitryl chloride production on air quality across the United States. Atmos. Chem. Phys. 2012, 12, 6455–6473. [Google Scholar] [CrossRef]
- EPA, 2021. 2017 National Emissions Inventory: January 2021 Updated Release, Technical Support Document, U.S. Environmental Protection Agency, Office of Air Quality Planning and Standards Air Quality Assessment Division, Emissions Inventory and Analysis Group, Research Triangle Park, NC, EPA-454/R-21-001, February 2021. Available online: https://www.epa.gov/sites/default/files/2021-02/documents/nei2017_tsd_full_jan2021.pdf (accessed on 2 March 2022).
- Gantt, B.; Kelly, J.T.; Bash, J.O. Updating sea spray aerosol emissions in the Community Multiscale Air Quality (CMAQ) Model Version 5.0.2. Geosci. Model Dev. 2015, 8, 3733–3746. Available online: www.geosci-model-dev.net/8/3733/2015/ (accessed on 15 March 2022). [CrossRef]
- Kang, D.; Pickering, K.E.; Allen, D.J.; Foley, K.M.; Wong, D.C.; Mathur, R.; Roselle, S.J. Simulating lightning NO production in CMAQv5.2: Evolution of scientific updates. Geosci. Model Dev. 2019, 12, 3071–3083. [Google Scholar] [CrossRef]
- Kettle, A.J.; Andreae, M.O.; Amouroux, D.; Andreae, T.W.; Bates, T.S.; Berresheim, H.; Bingemer, H.; Boniforti, R.; Curran, M.A.J.; DiTullio, G.R.; et al. A global database of sea surface dimethylsulfide (DMS) measurements and a procedure to predict sea surface DMS as a function of latitude, longitude, and month. Glob. Biogeochem. Cycles 1999, 13, 399–444. [Google Scholar] [CrossRef]
- Kettle, A.J.; Andreae, M.O. Flux of dimethylsulfide from the oceans: A comparison of updated data sets and flux models. J. Geophys. Res. Atmos. 2000, 105, 26793–26808. [Google Scholar] [CrossRef]
- Liss, P.S.; Merlivat, L. Air-Sea Gas Exchange Rates: Introduction and Synthesis. In The Role of Air-Sea Exchange in Geochemical Cycling; Buat-Ménard, P., Ed.; Springer: Dordrecht, The Netherlands, 1986; pp. 113–127. [Google Scholar]
- Nightingale, P.D.; Malin, G.; Law, C.S.; Watson, A.J.; Liss, P.S.; Liddicoat, M.I.; Boutin, J. Upstill-Goddard, R.C. In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers. Glob. Biogeochem. Cycles 2000, 14, 373–387. [Google Scholar] [CrossRef]
- Wanninkhof, R. Relationship between wind speed and gas exchange over the ocean. J. Geophys. Res. Ocean. 1992, 97, 7373–7382. [Google Scholar] [CrossRef]
- Saltzman, E.S.; King, D.B.; Holmen, K.; Leck, C. Experimental determination of the diffusion coefficient of dimethyl sulfide in water. J. Geophys. Res. Oceans 1993, 98, 16481–16486. [Google Scholar] [CrossRef]
- McGillis, W.R.; Dacey, J.; Frew, N.M.; Bock, E.J.; Nelson, R.K. Water-air flux of dimethylsulfide. J. Geophys. Res. Oceans 2000, 105, 1187–1193. [Google Scholar] [CrossRef]
- Smith, S.N.; Mueller, S.F. Modeling natural emissions in the Community Multiscale Air Quality (CMAQ) Model–I: Building an Emissions Data Base. Atmos. Chem. Phys. 2010, 10, 4931–4952. [Google Scholar] [CrossRef]
- Sander, S.; Friedl, R.; Abbatt, J.; Barker, J.; Burkholder, J.; Golden, D.; Kolb, C.; Kurylo, M.; Moortgat, G.; Wine, P.J.J. Chemical kinetics and photochemical data for use in atmospheric studies, evaluation number. JPL 2011, 14, 10. [Google Scholar]
- Atkinson, R.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Kerr, J.A.; Rossi, M.J.; Troe, J. Summary of Evaluated Kinetic and Photo-Chemical Data for Atmospheric Chemistry; Technical Report; IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry: October 2006. Available online: http://www.iupac-kinetic.ch.cam.ac.uk/ (accessed on 27 December 2019).
- Sommariva, R.; von Glasow, R. Multiphase Halogen Chemistry in the Tropical Atlantic Ocean. Environ. Sci. Technol. 2012, 46, 10429–10437. [Google Scholar] [CrossRef]
- Mathur, R.; Xing, J.; Gilliam, R.; Sarwar, G.; Hogrefe, C.; Pleim, J.; Pouliot, G.; Roselle, S.; Spero, T.L.; Wong, D.C.; et al. Extending the Community Multiscale Air Quality (CMAQ) modeling system to hemispheric scales: Overview of process considerations and initial applications. Atmos. Chem. Phys. 2017, 17, 12449–12474. [Google Scholar] [CrossRef]
- Lelieveld, J.; Roelofs, G.-J.; Ganzeveld, L.; Feichter, J.; Rodhe, H. Terrestrial sources and distribution of atmospheric sulphur. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 1997, 352, 149–158. [Google Scholar] [CrossRef]
- Hulswar, S.; Simó, R.; Galí, M.; Bell, T.G.; Lana, A.; Inamdar, S.; Halloran, P.R.; Manville, G.; Mahajan, A.S. Third revision of the global surface seawater dimethyl sulfide climatology (DMS-Rev3). Earth Syst. Sci. Data 2022, 14, 2963–2987. [Google Scholar] [CrossRef]
- Veres, P.R.; Neuman, J.A.; Bertram, T.H.; Assaf, E.; Wolfe, G.M.; Williamson, C.J.; Weinzierl, B.; Tilmes, S.; Thompson, C.R.; Thames, A.B.; et al. Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere. Proc. Natl. Acad. Sci. USA 2020, 117, 4505. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.A.; Fite, C.H.; Holmes, C.D.; Veres, P.R.; Neuman, J.A.; Faloona, I.; Thornton, J.A.; Wolfe, G.M.; Vermeuel, M.P.; Jernigan, C.M.; et al. Rapid cloud removal of dimethyl sulfide oxidation products limits SO2 and cloud condensation nuclei production in the marine atmosphere. Proc. Natl. Acad. Sci. USA 2021, 118, e2110472118. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Reaction | Rate Expression (cm3 molecule−1 s−1) | References |
|---|---|---|---|
| 1 | DMS + OH = SO2 +… (abstraction channel) | k = 1.12 × 10−11 e−250/T T = temperature in Kelvin | [53] |
| 2 | DMS + OH = 0.75 × SO2 +… (addition channel) | ko = 1.99 × 10−39 e−5270/T k∞ = 1.26 × 10−10 e+340/T k = {ko[M]/(1 + ko[M]/k∞)} Fz Z = {(1/N) + log10[ko [M]/k∞]2}−1 F = 1.0 and N = 1.0 [M] = total pressure, molecules/cm3 | [53] |
| 3 | DMS + NO3 = SO2 +… | k = 1.93 × 10−13 e+520/T | [53] |
| 4 | DMS + Cl = 0.86 × SO2 +… | k = 3.4 × 10−13 e+2081/T | [54,55] |
| Studies | Enhancements µg/m3 | Sources Considered | Geographic Region | Season |
|---|---|---|---|---|
| Park et al. [13] | Ammonium sulfate 0.11 | DMS from seawater, volcanoes, and biomass burning activities | Eastern and western US | Annual |
| This study | Ammonium sulfate 0.07 | DMS from seawater | Entire land area of the modeling domain | Annual |
| Mueller et al. [18] | Ammonium sulfate 0.12 | DMS and hydrogen sulfide from seawater, coastal wetlands, freshwater, Great Salt Lake, soils, volcanoes, and fumaroles | Entire modeling domain | Winter (December–February) |
| This study | Ammonium sulfate 0.08 | DMS from seawater | Entire modeling domain | Winter (December–February) |
| Mueller et al. [18] | Ammonium sulfate 0.27 | DMS and hydrogen sulfide from seawater, coastal wetlands, freshwater, Great Salt Lake, soils, volcanoes, and fumaroles | Entire modeling domain | Summer (June–August) |
| This study | Ammonium sulfate 0.18 | DMS from seawater | Entire modeling domain | Summer (June–August) |
| Zhao et al. [20] | Sulfate 0.08 | DMS from seawater | Entire US | Annual |
| This study | Sulfate 0.055 | DMS from seawater | Entire US | Annual |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarwar, G.; Kang, D.; Henderson, B.H.; Hogrefe, C.; Appel, W.; Mathur, R. Examining the Impact of Dimethyl Sulfide Emissions on Atmospheric Sulfate over the Continental U.S. Atmosphere 2023, 14, 660. https://doi.org/10.3390/atmos14040660
Sarwar G, Kang D, Henderson BH, Hogrefe C, Appel W, Mathur R. Examining the Impact of Dimethyl Sulfide Emissions on Atmospheric Sulfate over the Continental U.S. Atmosphere. 2023; 14(4):660. https://doi.org/10.3390/atmos14040660
Chicago/Turabian StyleSarwar, Golam, Daiwen Kang, Barron H. Henderson, Christian Hogrefe, Wyat Appel, and Rohit Mathur. 2023. "Examining the Impact of Dimethyl Sulfide Emissions on Atmospheric Sulfate over the Continental U.S." Atmosphere 14, no. 4: 660. https://doi.org/10.3390/atmos14040660
APA StyleSarwar, G., Kang, D., Henderson, B. H., Hogrefe, C., Appel, W., & Mathur, R. (2023). Examining the Impact of Dimethyl Sulfide Emissions on Atmospheric Sulfate over the Continental U.S. Atmosphere, 14(4), 660. https://doi.org/10.3390/atmos14040660