
Theoretical Perspectives on the Gas-Phase Oxidation Mechanism and Kinetics of Carbazole Initiated by OH Radical in the Atmosphere

Abstract
:1. Introduction
2. Computational Detail
2.1. Thermodynamic Calculation
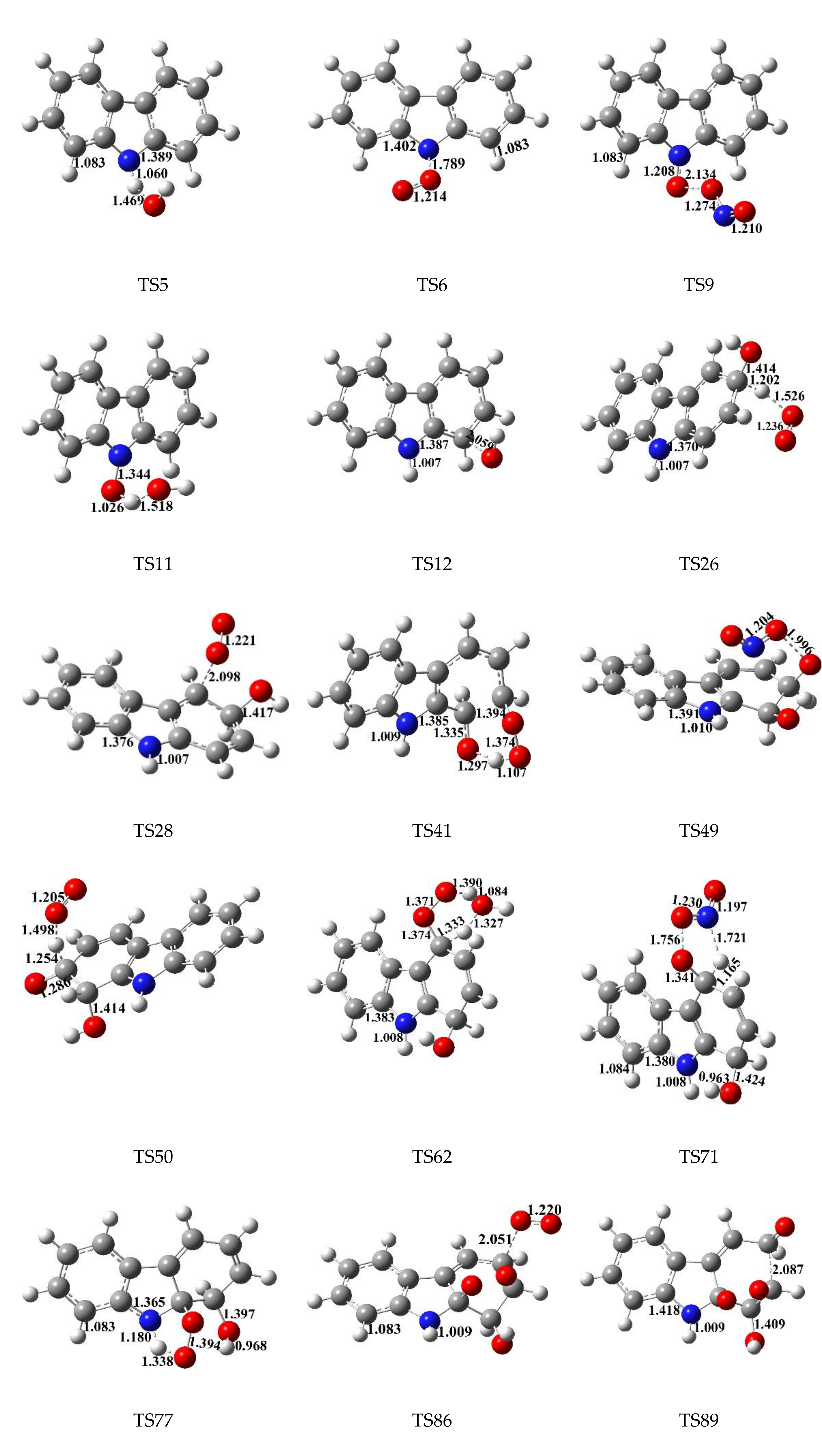
2.2. Kinetic Calculation
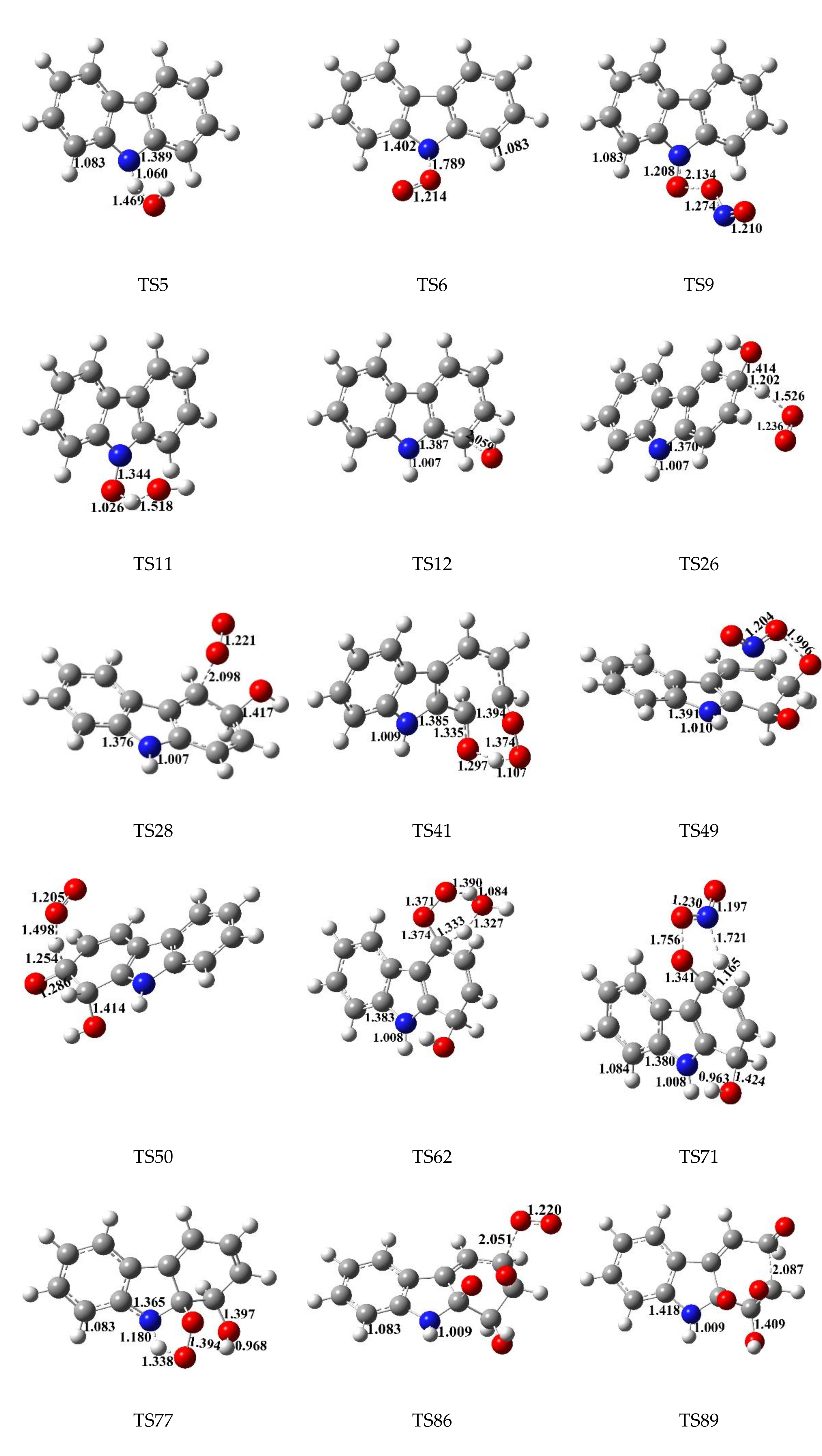
3. Results and Discussion
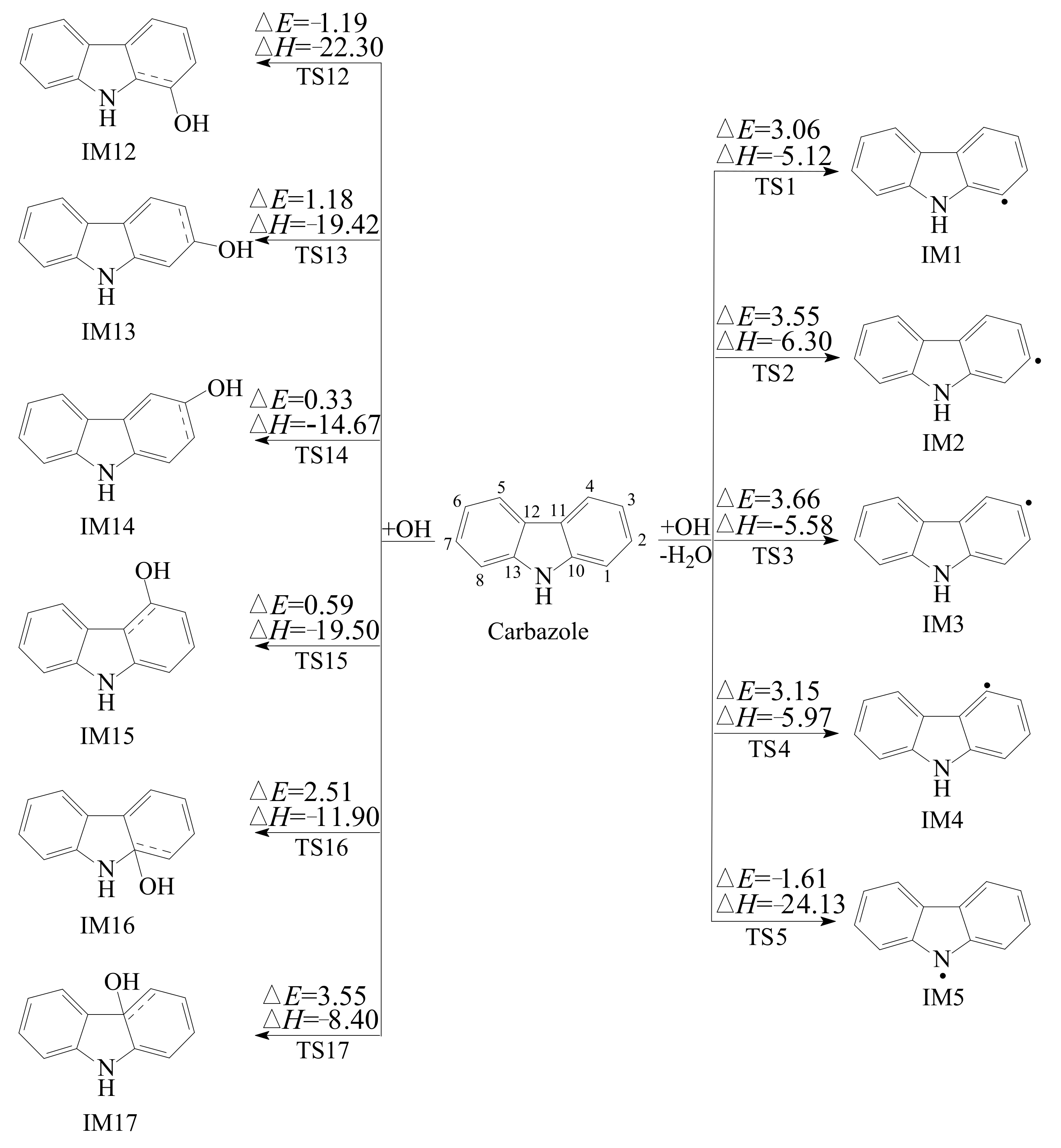
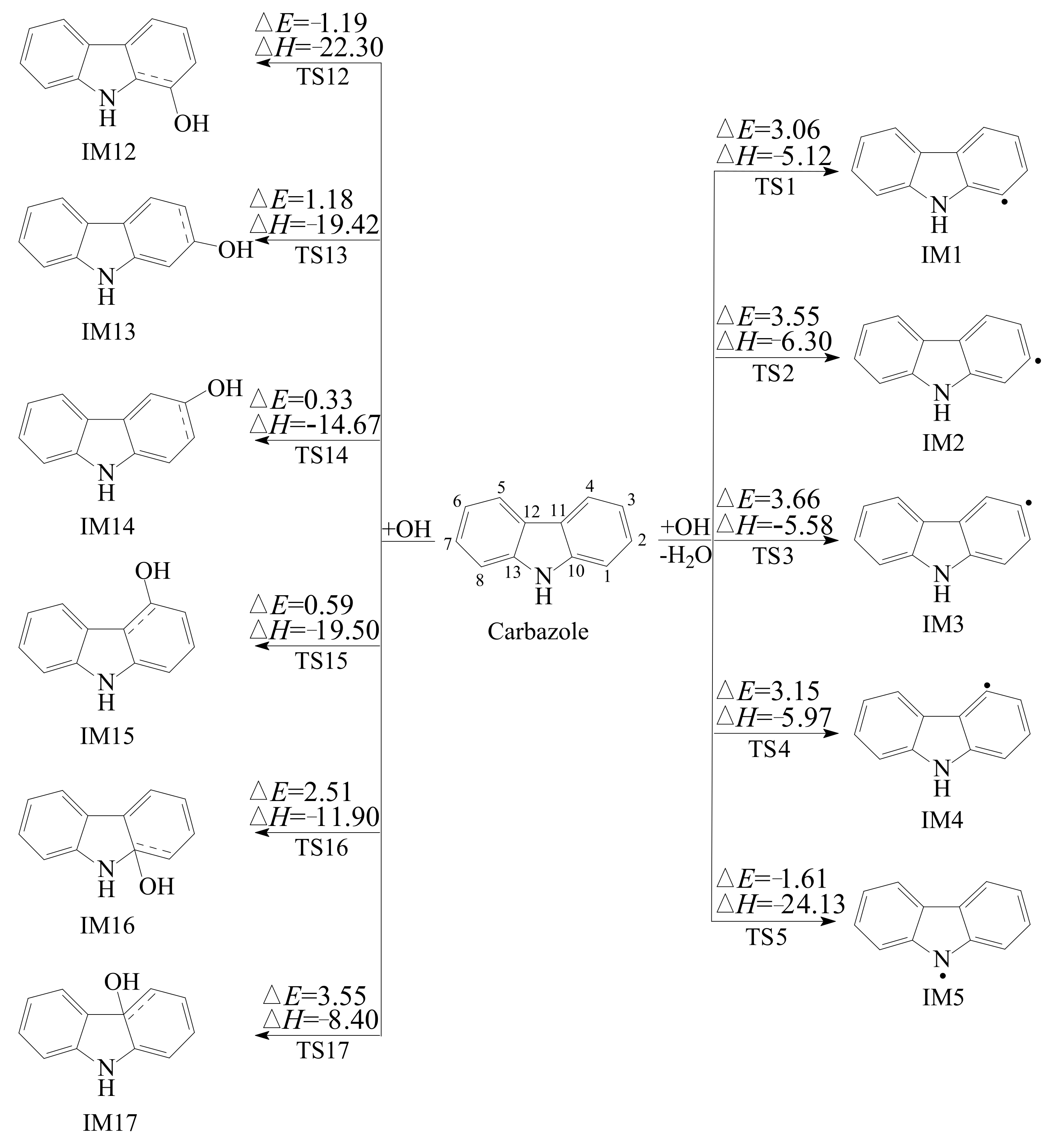
3.1. Reaction of Carbazole and OH Radical
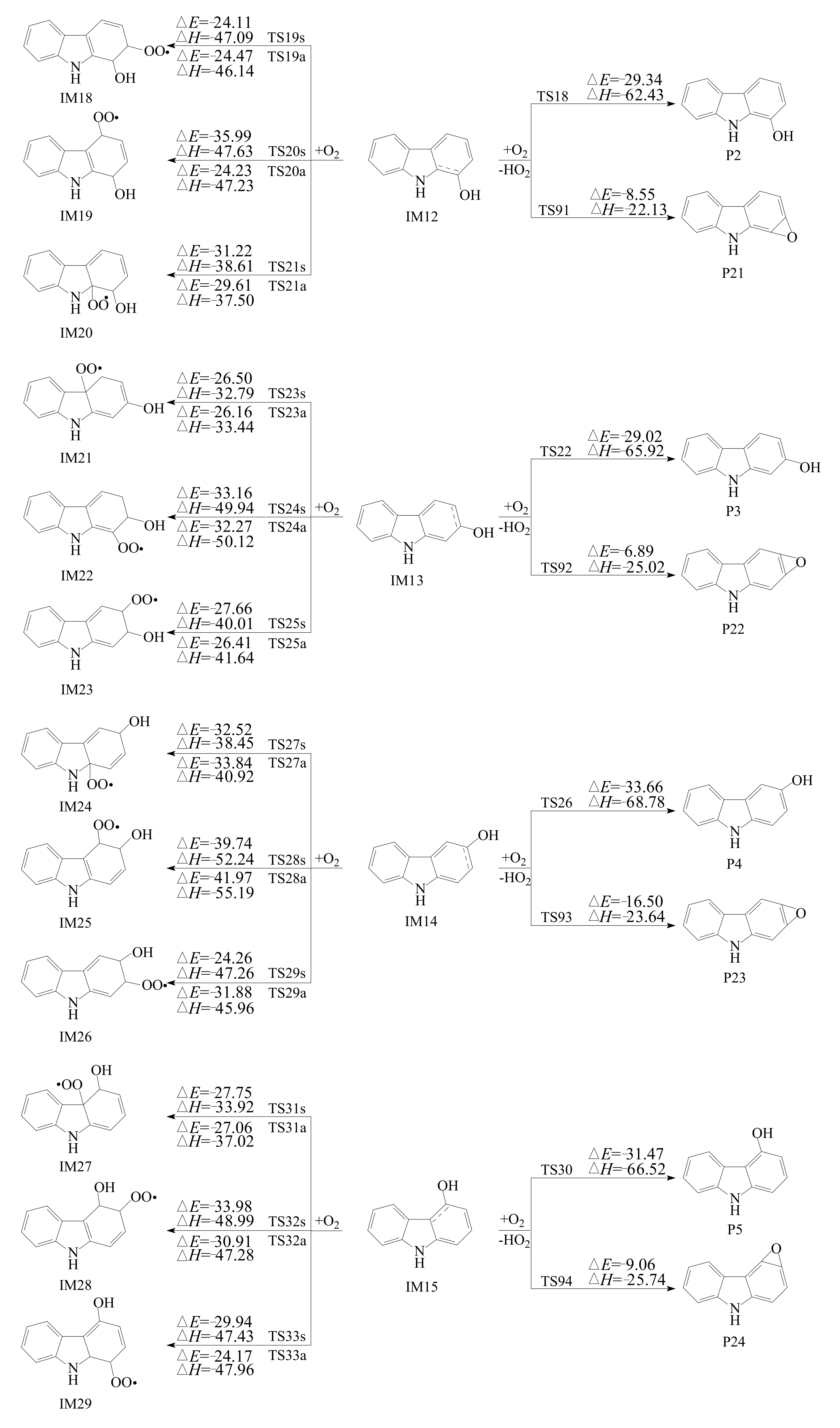
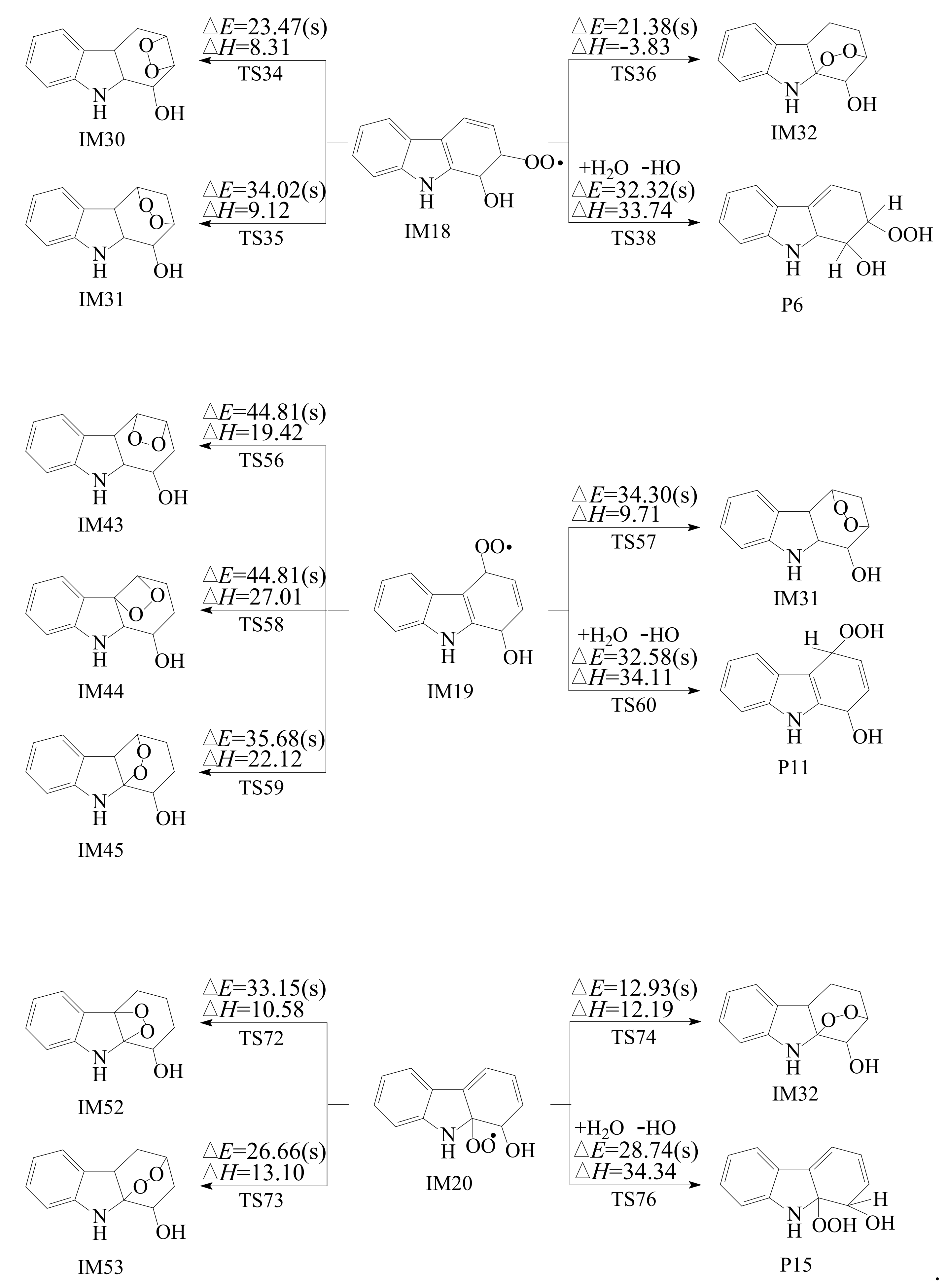
3.2. Subsequent Reaction
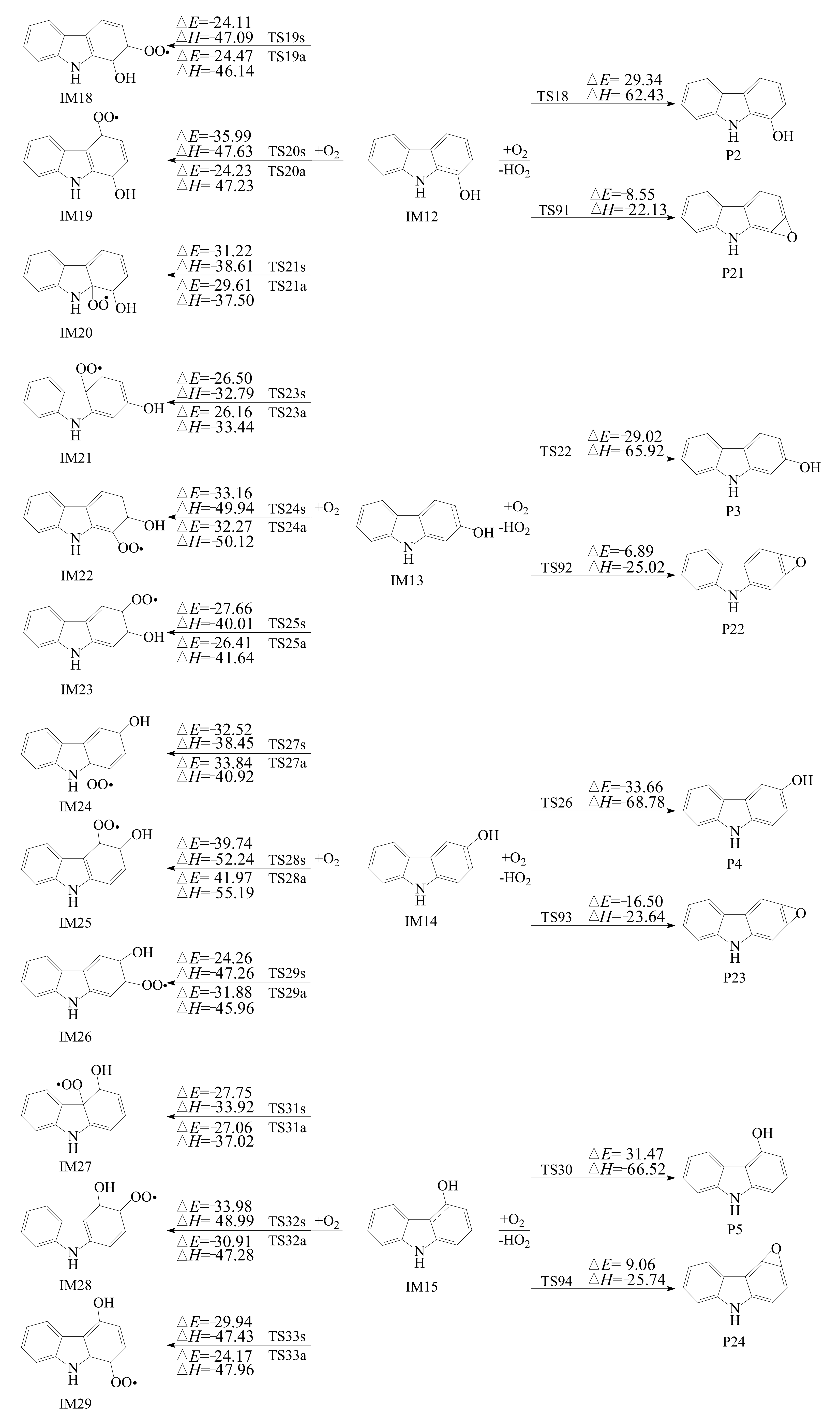
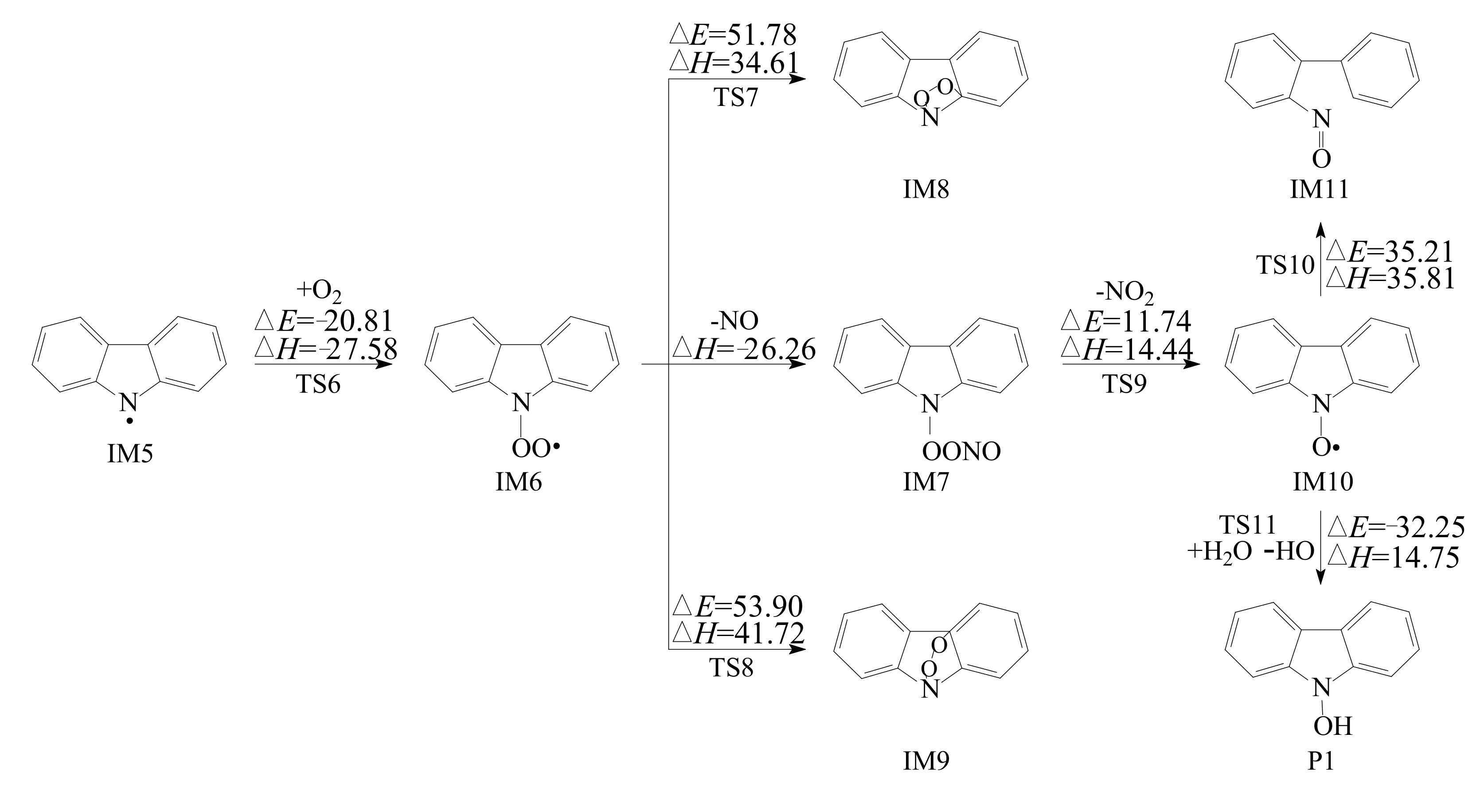
3.2.1. Reactions with O2
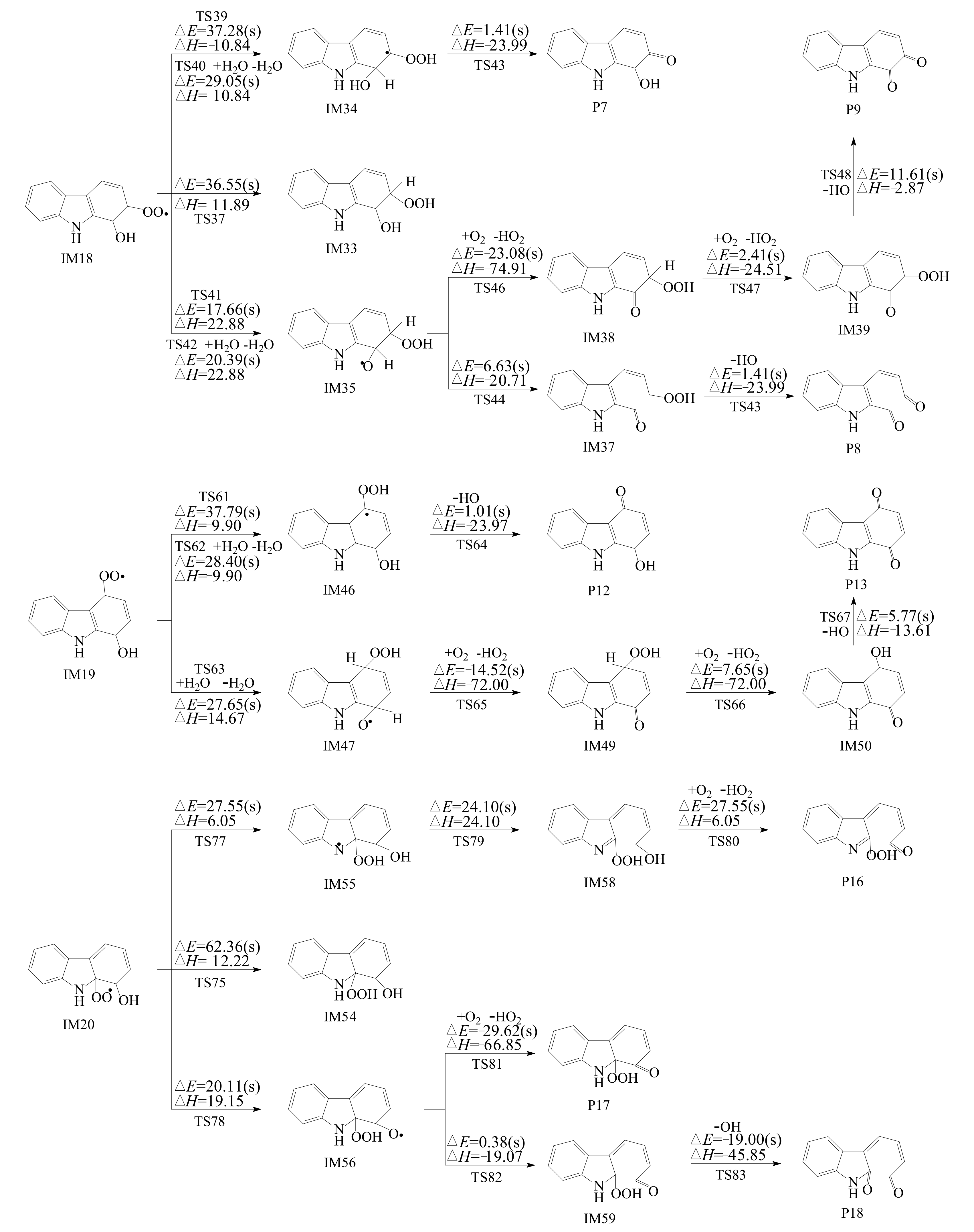
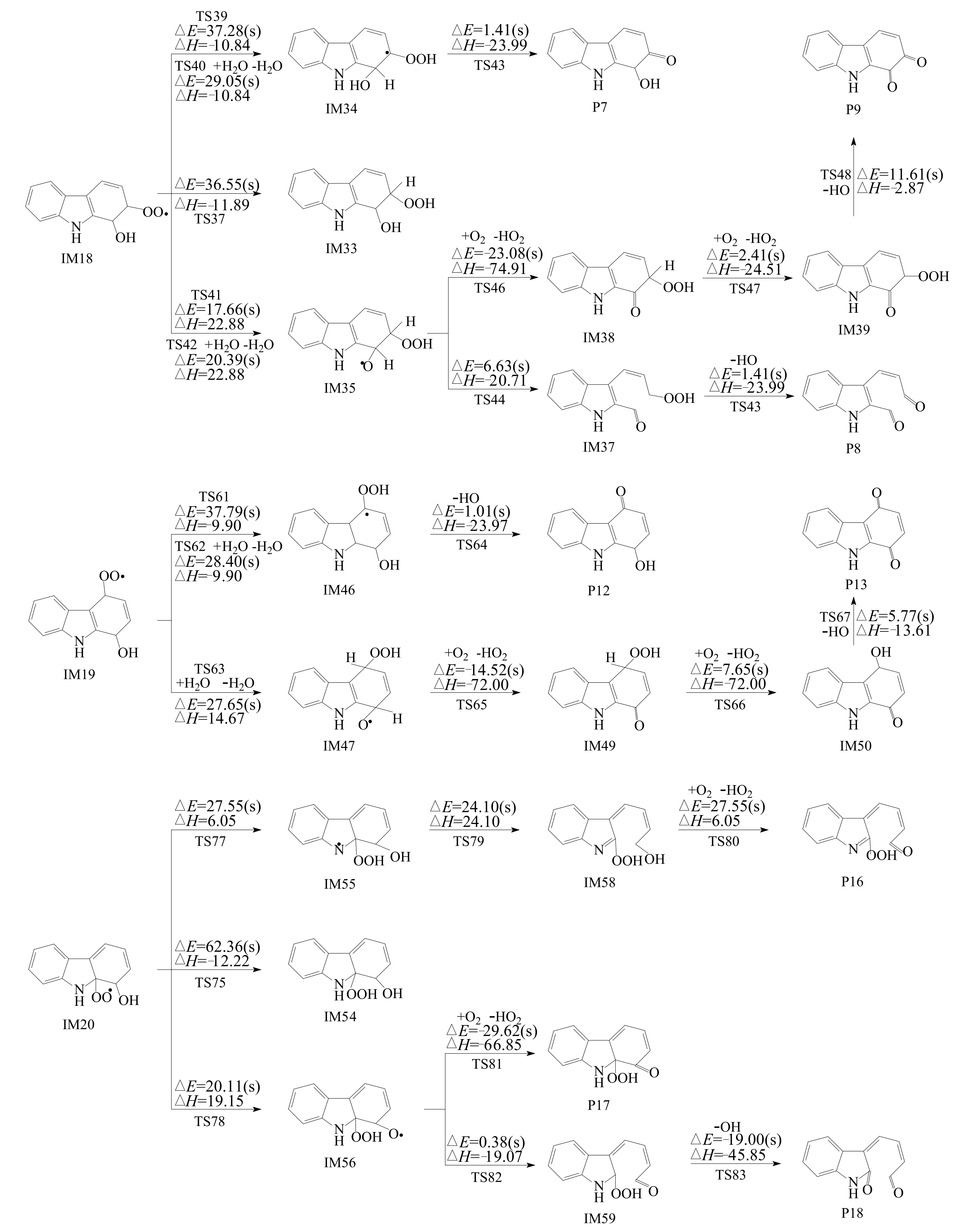
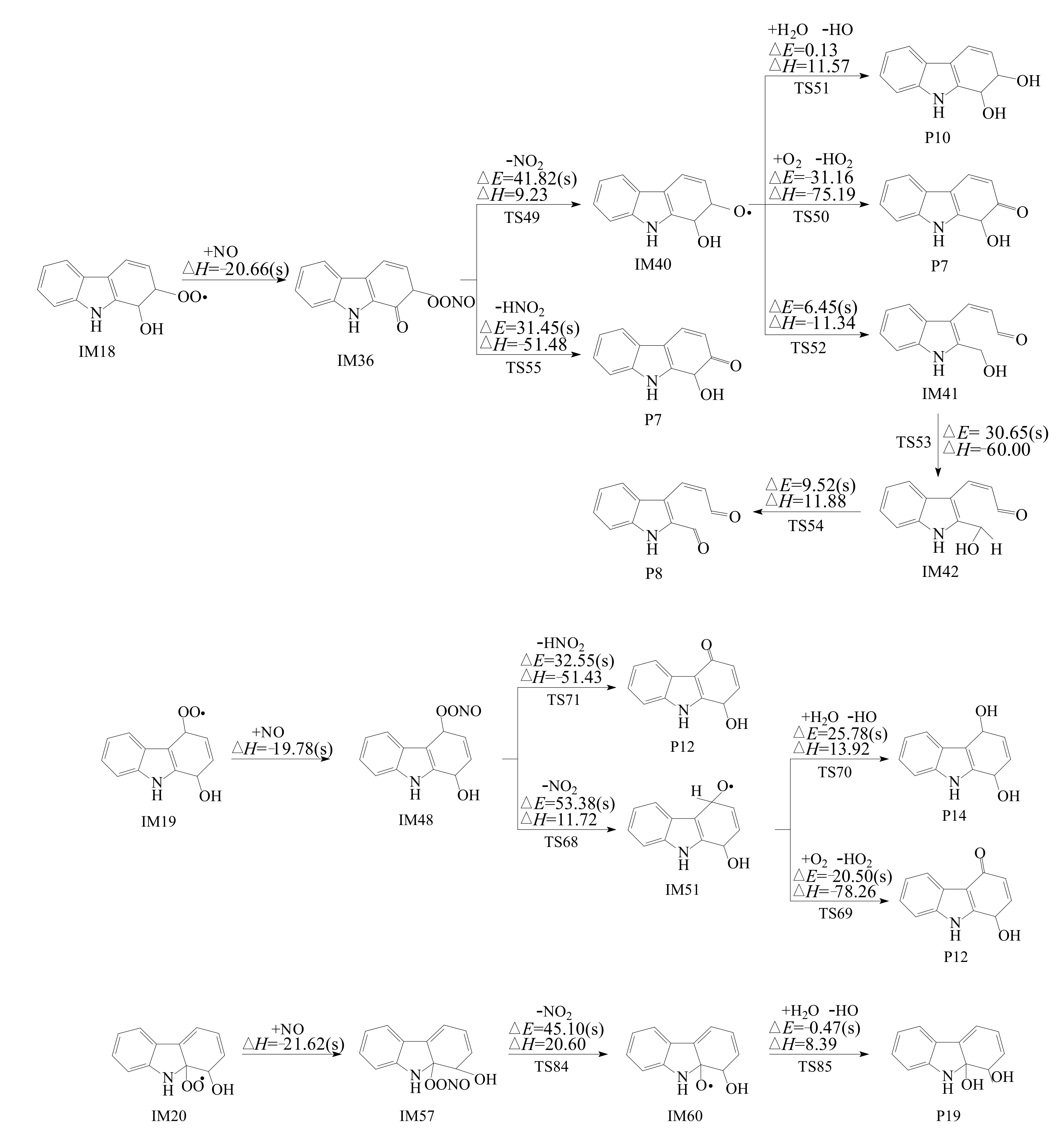
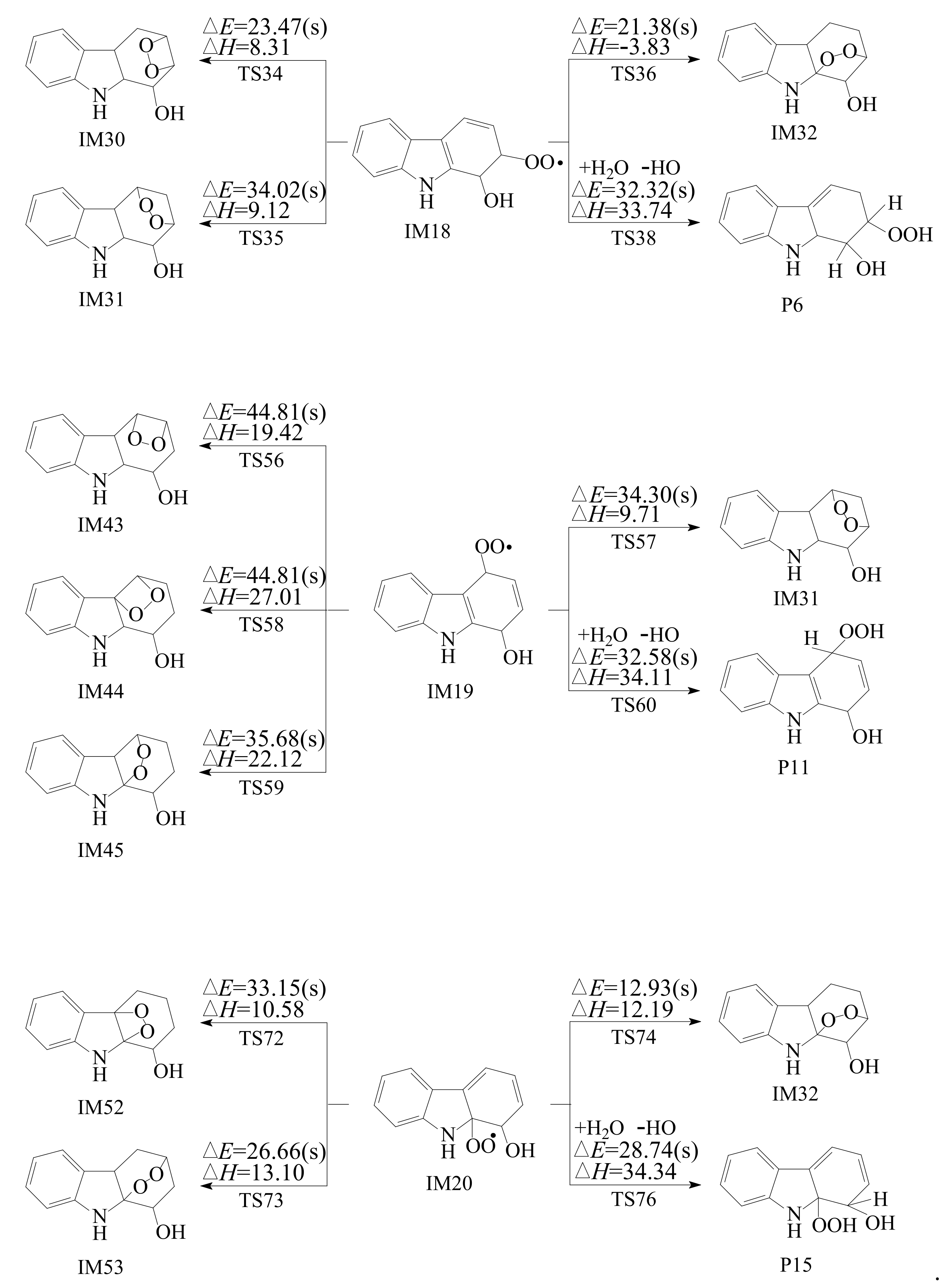
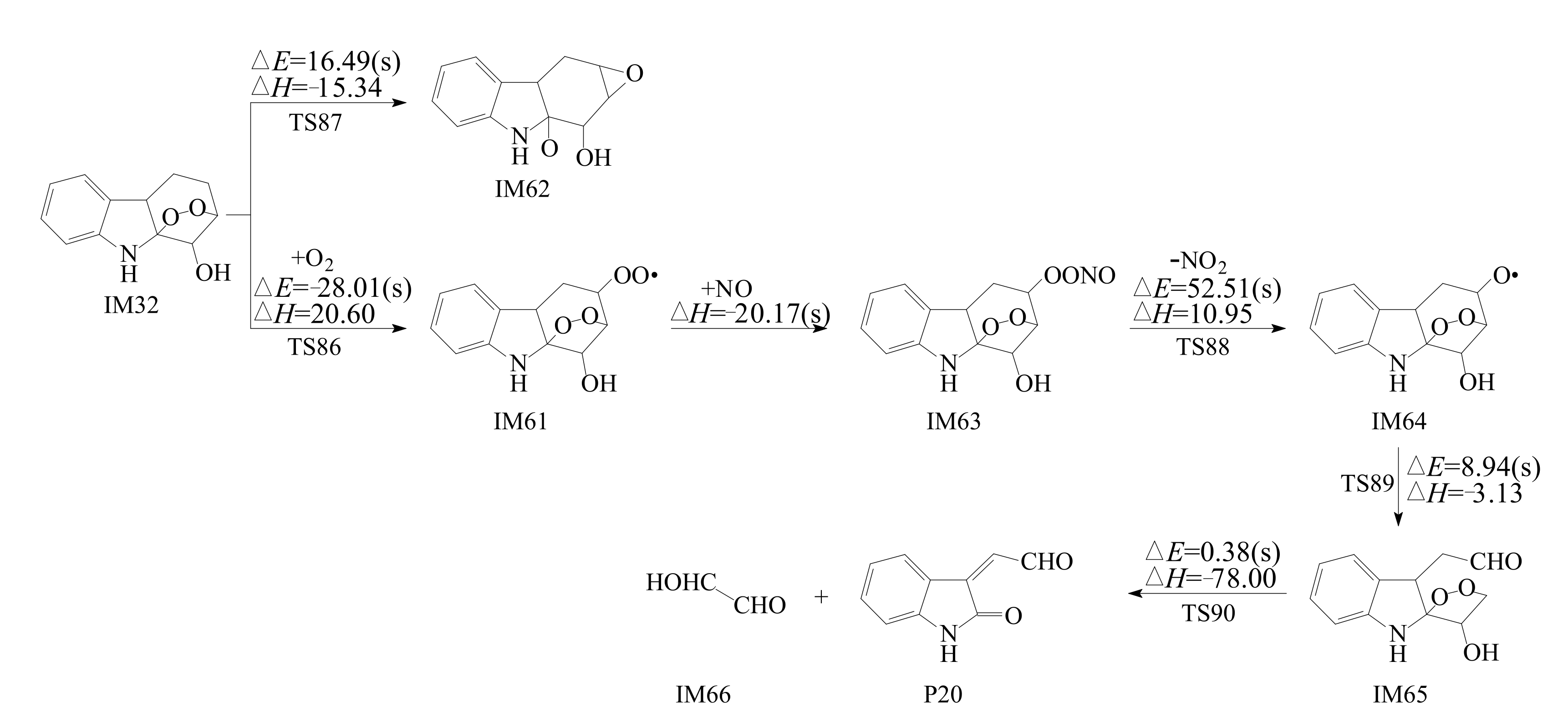
3.2.2. Subsequent Reaction of Carbazole-OH-O2 Intermediate
- (1)
- Intramolecular H-transfer reaction
- (2)
- NO addition reaction
- (3)
- Reactions of bicyclic peroxy radicals
3.3. Rate Constant Calculations
4. Conclusions
- There are four types of reactions for the degradation of carbazole initiated by OH radical: OH additions to “bend” C atoms, OH additions to “benzene ring” C atoms, H abstractions from C-H bonds, and the H abstraction from N-H bond. Among them, OH additions to “bend” C atoms and H abstractions from C-H bonds are energetically unfavorable. The best pathway of OH addition reactions is OH addition to C1 atom, which is competitive with the H abstraction from the N-H bond.
- The primary products of carbazole oxidation by OH radicals in the atmosphere include hydroxycarbazole, dialdehyde, carbazolequinone, carbazole-ol, hydroxy-carbazole-one, and hydroperoxyl-carbazole-one.
- The degradation of carbazole in the atmosphere is significant, for which the rate constant determined by OH radical is 6.52 × 10−12 cm3 molecule−1 s−1 and the lifetime is 37.70 h. The OH addition on C1 and H abstraction on N atom account for 94% of the total reaction rate. The ranking of the rate constant for the reaction of NSO-HETs with OH is as follows: Carbazole ≈ Dibenzothiophene < Dibenzofuran ≈ Fluorene < Pyrrole ≈ Indole.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Johansen, S.S.; Hansen, A.B.; Mosbaek, H.; Arvin, E. Identification of Heteroaromatic and Other Organic Compounds in Ground Water at Creosote-Contaminated sites in Denmark. Ground Water Monit. Remediat. 1997, 17, 106–115. [Google Scholar] [CrossRef]
- World Health Organization. Iarc Monographs on the Evaluation of Carcinogenic Risks to Humans Preamble. Monogr. Eval. Carcinog. Risks Hum. 2008, 97, 9–38. [Google Scholar]
- Tovar, C.M.; Barnes, I.; Bejan, I.G.; Wiesen, P. Kinetic study of the atmospheric oxidation of a series of epoxy compounds by OH radicals. Atmos. Chem. Phys. 2022, 22, 6989–7004. [Google Scholar] [CrossRef]
- Eisentraeger, A.; Brinkmann, C.; Hollert, H.; Sagner, A.; Tiehm, A.; Neuwoehner, J. Heterocyclic Compounds: Toxic Effects Using Algae, Daphnids, and the Salmonella/Microsome Test Taking Methodical Quantitative Aspects into Account. Environ. Toxicol. Chem. 2008, 27, 1590–1596. [Google Scholar] [CrossRef]
- Esen, F.; Tasdemir, Y.; Cindoruk, S.S. Dry Deposition, Concentration and Gas/Particle Partitioning of Atmospheric Carbazole. Atmos. Res. 2010, 95, 379–385. [Google Scholar] [CrossRef]
- Roy, J.; Jana, A.K.; Mal, D. Recent Trends in the Synthesis of Carbazoles: An Update. Tetrahedron 2012, 68, 6099–6121. [Google Scholar] [CrossRef]
- Ishikawa, S.; Sakazaki, Y.; Eguchi, Y.; Suetomi, R.; Nakamura, E. Identification of Chemical Substances in Industrial Wastes and Their Pyrolytic Decomposition Products. Chemosphere 2005, 59, 1343–1353. [Google Scholar] [CrossRef]
- Glarborg, P.; Jensen, A.D.; Johnsson, J.E. Fuel Nitrogen Conversion in Solid Fuel Fired Systems. Prog. Energ. Combust. 2003, 29, 89–113. [Google Scholar] [CrossRef]
- Fromme, H.; Mi, W.; Lahrz, T.; Kraft, M.; Aschenbrenner, B.; Bruessow, B.; Ebinghaus, R.; Xie, Z.; Fembacher, L. Occurrence of Carbazoles in Dust and Air Samples from Different Locations in Germany. Sci. Total Environ. 2018, 610–611, 412–418. [Google Scholar] [CrossRef]
- Parette, R.; McCrindle, R.; McMahon, K.S.; Pena-Abaurrea, M.; Reiner, E.; Chittim, B.; Riddell, N.; Voss, G.; Dorman, F.L.; Pearson, W.N. Halogenated Indigo Dyes: A Likely Source of 1,3,6,8-Tetrabromocarbazole and Some Other Halogenated Carbazoles in the Environment. Chemosphere 2015, 127, 18–26. [Google Scholar] [CrossRef]
- Campbell, N.; Barclay, B.M. Recent Advances in the Chemistry of Carbazole. Chem. Rev. 1947, 40, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.M.; Kara, M.; Dumanoglu, Y.; Odabasi, M.; Elbir, T. Source Apportionment of Polycyclic Aromatic Hydrocarbons (PAHs) and Polychlorinated Biphenyls (PCBs) in Ambient Air of an Industrial Region in Turkey. Atmos. Environ. 2014, 97, 271–285. [Google Scholar] [CrossRef]
- Shi, S.N.; Qu, Y.Y.; Ma, F.; Zhou, J.T. Bioremediation of Coking Wastewater Containing Carbazole, Dibenzofuran and Dibenzothiophene by Immobilized Naphthalene-Cultivated Arthrobacter sp. W1 in Magnetic Gellan Gum. Bioresour. Technol. 2014, 166, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Yu, B.; Li, F.L.; Cai, X.F.; Ma, C.Q. Microbial Degradation of Sulfur, Nitrogen and Oxygen Heterocycles. Trends Microbiol. 2006, 14, 398–405. [Google Scholar] [CrossRef]
- Larentis, A.L.; Sampaio, H.C.C.; Carneiro, C.C.; Martins, O.B.; Alves, T.L.M. Evaluation of Growth, Carbazole Biodegradation and Anthranilic Acid Production by Pseudomonas Stutzeri. Braz. J. Chem. Eng. 2011, 28, 37–44. [Google Scholar] [CrossRef]
- Nojiri, H.; Omori, T. Carbazole Metabolism by Pseudomonads. In Pseudomonas: A Model System in Biology; Filloux, A., Ed.; Springer: Dordrecht, The Netherlands, 2007; pp. 107–145. [Google Scholar]
- Heim, S.; Schwarzbauer, J.; Kronimus, A.; Littke, R.; Woda, C.; Mangini, A. Geochronology of Anthropogenic Pollutants in Riparian Wetland Sediments of the Lippe River (Germany). Org. Geochem. 2004, 35, 1409–1425. [Google Scholar] [CrossRef]
- Lelieveld, J.; Gromov, S.; Pozzer, A.; Taraborrelli, D. Global tropospheric hydroxyl distribution, budget and reactivity. Atmos. Chem. Phys. 2016, 16, 12477–12493. [Google Scholar] [CrossRef] [Green Version]
- Keyte, I.J.; Harrison, R.M.; Lammel, G. Chemical Reactivity and Long-Range Transport Potential of Polycyclic Aromatic Hydrocarbons-A Review. Chem. Soc. Rev. 2013, 42, 9333–9391. [Google Scholar] [CrossRef]
- Kurylo, M.J.; Orkin, V.L. Determination of Atmospheric Lifetimes via the Measurement of OH Radical Kinetics. Chem. Rev. 2003, 103, 5049–5076. [Google Scholar] [CrossRef]
- Wang, S.; Chen, D. Study an the Generated Mechanism of Carbazole Nitroxyl Radical via Photocatalysis Oxidation of Carbazole. Acta Phys. Chim. Sin. 1995, 11, 1014–1019. [Google Scholar]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current Status of Transition-State Theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Rice, O.K.; Ramsperger, H.C. Theories of Unimolecular Gas Reactions at Low Pressures. J. Am. Chem. Soc. 1927, 49, 1617–1629. [Google Scholar] [CrossRef]
- Altarawneh, M.; Kennedy, E.M.; Dlugogorski, B.Z.; Mackie, J.C. Computational Study of the Oxidation and Decomposition of Dibenzofuran Under Atmospheric Conditions. J. Phys. Chem. A 2008, 112, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.V.; Nguyen, H.T.; Huynh, L.K. Ab initio Kinetic Mechanism of OH-initiated Atmospheric Oxidation of Pyrrole. Chemosphere 2021, 263, 127850. [Google Scholar] [CrossRef]
- Ding, Z.Z.; Yi, Y.Y.; Zhang, Q.Z.; Zhuang, T. Theoretical Investigation on Atmospheric Oxidation of Fluorene Initiated by OH Radical. Sci. Total Environ. 2019, 669, 920–929. [Google Scholar] [CrossRef]
- Kwok, E.S.C.; Atkinson, R.; Arey, J. Kinetics of the Gas-phase Reactions of Dibenzothiophene with OH radicals, NO3 Radicals, and O3. Polycycl. Aromat. Comp. 1999, 13, 175–189. [Google Scholar] [CrossRef]
- Wallington, T.J. Kinetics of the gas-phase reaction of OH radicals with Pyrrole and Thiophene. Int. J. Chem. Kinet. 1986, 18, 487–496. [Google Scholar] [CrossRef]
- Atkinson, R.; Tuazon, E.C.; Arey, J.; Aschmann, S.M. Atmospheric and Indoor Chemistry of Gas-phase Indole, Quinoline, and Isoquinoline. Atmos. Environ. 1995, 29, 3423–3432. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09W, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Fukui, K. The Path of Chemical Reactions-The IRC Approach. Accounts Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Glowacki, D.R.; Liang, C.H.; Morley, C.; Pilling, M.J.; Robertson, S.H. Mesmer: An Open-source Master Equation Solver for Multi-energy Well Reactions. J. Phys. Chem. A 2012, 116, 9545–9560. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.G.; Smith, S.C. Theory of Unimolecular and Recombination Reactions; Blackwell Science Inc.: Hoboken, NY, USA, 1990; pp. 341–350. [Google Scholar]
- Lee, S.Y.; Boo, B.H. Molecular Structures and Vibrational Spectra of Pyrrole and Carbazole by density Functional Theory and Conventional Ab Initio Calculations. J. Phys. Chem. 1996, 100, 15073–15078. [Google Scholar] [CrossRef]
- Batra, I.P.; Bagus, P.S.; Clementi, E.; Seki, H. Ab-initio Calculations for Electronic-structure of Carbazole and Trinitrofluorenone. Theoret. Chim. Acta 1974, 32, 279–293. [Google Scholar] [CrossRef]
- Lorenz, K.; Zellner, R. Kinetics of the reactions of OH-radicals with benzene, benzene-d6 and naphthalene. Ber. Bunsenges. Phys. Chem. 1983, 87, 629–636. [Google Scholar] [CrossRef]
- Ananthula, R.; Yamada, T.; Taylor, P.H. Kinetics of OH Radical Reaction with Anthracene and Anthracene-d10. J. Phys. Chem. A 2006, 110, 3559–3566. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Stevens, P.S.; Hites, R.A. Rate Constants for the Gas-phase Reactions of Methylphenanthrenes with OH as a Function of Temperature. J. Phys. Chem. A 2003, 107, 6603–6608. [Google Scholar] [CrossRef]
- Ananthula, R.; Yamada, T.; Taylor, P.H. Kinetics of OH Radical Reaction with Phenanthrene: New Absolute Rate Measurements and Comparison with Other PAHs. Int. J. Chem. Kinet. 2007, 39, 629–637. [Google Scholar] [CrossRef]
- Dang, J.; Zhang, Q.Z. Gas-phase Reaction of Benzo a Anthracene with Hydroxyl Radical in the Atmosphere: Products, Oxidation Mechanism, and Kinetics. J. Mol. Model. 2018, 24, 320. [Google Scholar] [CrossRef]
- Dang, J.; Shi, X.L.; Zhang, Q.Z.; Wang, W.X. Theoretical Perspectives on the Mechanism and Kinetics of the OH Radical-initiated Gas-phase oxidation of PCB126 in the Atmosphere. Sci. Total Environ. 2015, 517, 1–9. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, Q.; Wang, W. Atmospheric Oxidation of Phenanthrene Initiated by OH Radicals in the Presence of O2 and NOx-A Theoretical Study. Sci. Total Environ. 2016, 563, 1008–1015. [Google Scholar] [CrossRef]
- Jenkin, M.E.; Valorso, R.; Aumont, B.; Rickard, A.R.; Wallington, T.J. Estimation of Rate Coefficients and Branching Ratios for Gas-phase Reactions of OH with Aromatic Organic Compounds for Use in Automated Mechanism Construction. Atmos. Chem. Phys. 2018, 18, 9329–9349. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactions | Rate Constants |
|---|---|
| carbazole + OH (k) | 8.81 × 10−12 |
| carbazole + OH (kabs) | 3.70 × 10−12 |
| carbazole + OH → IM1 +H2O (k1) | 1.31 × 10−16 |
| carbazole + OH → IM2 + H2O (k2) | 2.03 × 10−16 |
| carbazole + OH → IM3 + H2O (k3) | 1.05 × 10−16 |
| carbazole + OH → IM4 + H2O (k4) | 2.03 × 10−16 |
| carbazole + OH → IM5 + H2O (k5) | 3.70 × 10−12 |
| carbazole + OH (kadd) | 2.82 × 10−12 |
| carbazole + OH → IM12 (k6) | 1.23 × 10−12 |
| carbazole + OH → IM13 (k7) | 2.10 × 10−14 |
| carbazole + OH → IM14 (k8) | 9.65 × 10−14 |
| carbazole + OH → IM15 (k9) | 6.09 × 10−14 |
| carbazole + OH → IM16 (k10) | 1.01 × 10−15 |
| carbazole + OH → IM17 (k11) | 1.61 × 10−16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, Z.; Wang, X.; Hadizadeh, M.H.; Han, Y.; Zhao, X.; Zhang, Q.; Wang, H.; Li, Y.; Xu, F.; Sun, Y. Theoretical Perspectives on the Gas-Phase Oxidation Mechanism and Kinetics of Carbazole Initiated by OH Radical in the Atmosphere. Atmosphere 2022, 13, 1129. https://doi.org/10.3390/atmos13071129
Teng Z, Wang X, Hadizadeh MH, Han Y, Zhao X, Zhang Q, Wang H, Li Y, Xu F, Sun Y. Theoretical Perspectives on the Gas-Phase Oxidation Mechanism and Kinetics of Carbazole Initiated by OH Radical in the Atmosphere. Atmosphere. 2022; 13(7):1129. https://doi.org/10.3390/atmos13071129
Chicago/Turabian StyleTeng, Zhuochao, Xiaotong Wang, Mohammad Hassan Hadizadeh, Yanan Han, Xianwei Zhao, Qi Zhang, Hetong Wang, Ying Li, Fei Xu, and Yanhui Sun. 2022. "Theoretical Perspectives on the Gas-Phase Oxidation Mechanism and Kinetics of Carbazole Initiated by OH Radical in the Atmosphere" Atmosphere 13, no. 7: 1129. https://doi.org/10.3390/atmos13071129
APA StyleTeng, Z., Wang, X., Hadizadeh, M. H., Han, Y., Zhao, X., Zhang, Q., Wang, H., Li, Y., Xu, F., & Sun, Y. (2022). Theoretical Perspectives on the Gas-Phase Oxidation Mechanism and Kinetics of Carbazole Initiated by OH Radical in the Atmosphere. Atmosphere, 13(7), 1129. https://doi.org/10.3390/atmos13071129