
Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides


Abstract
:1. Introduction
2. Experimental Methods
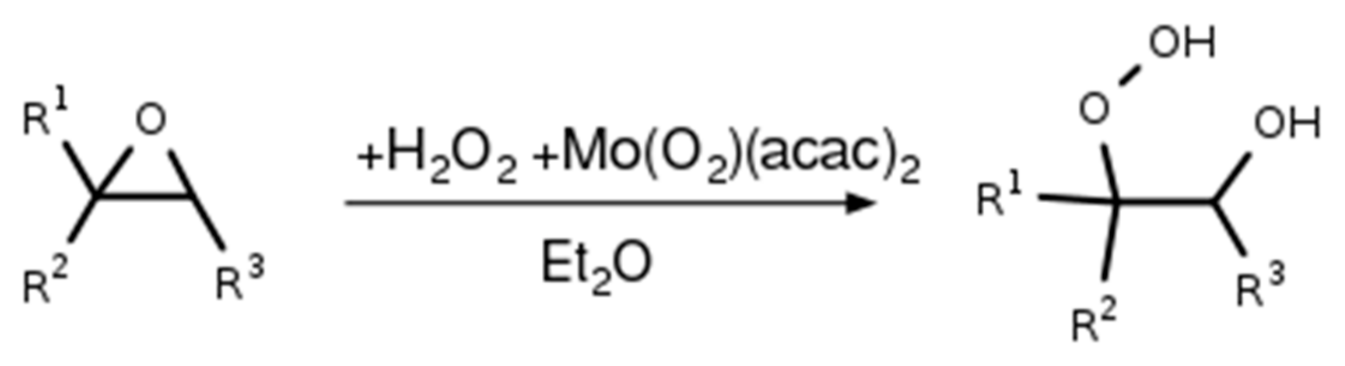
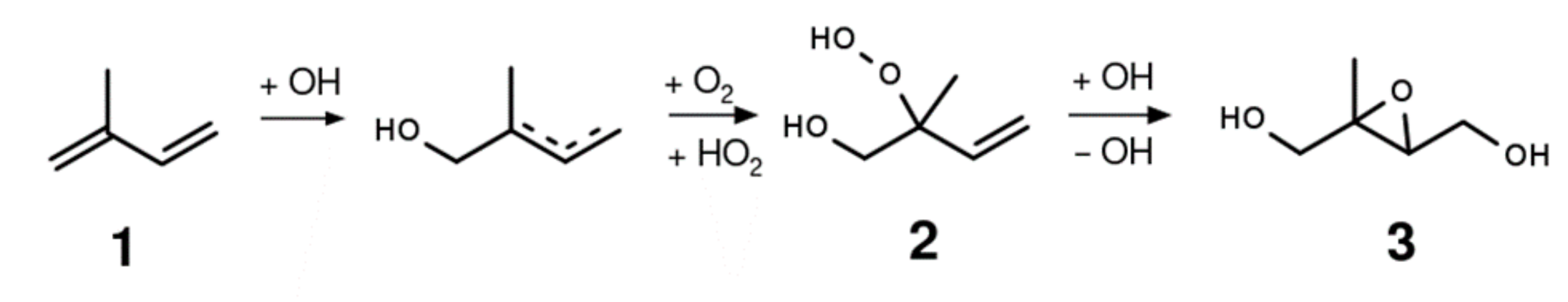
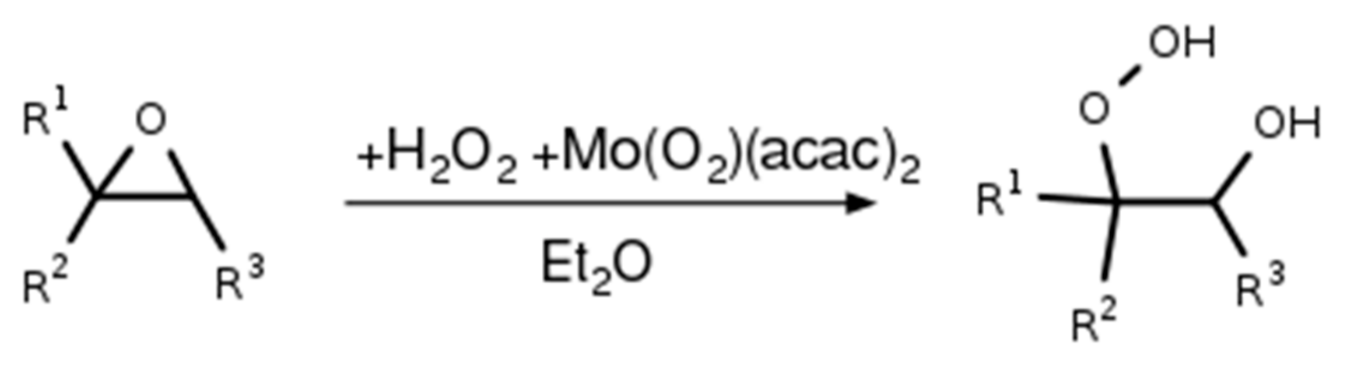
2.1. Synthesis of the Isoprene-Derived β-Hydroxy Hydroperoxides
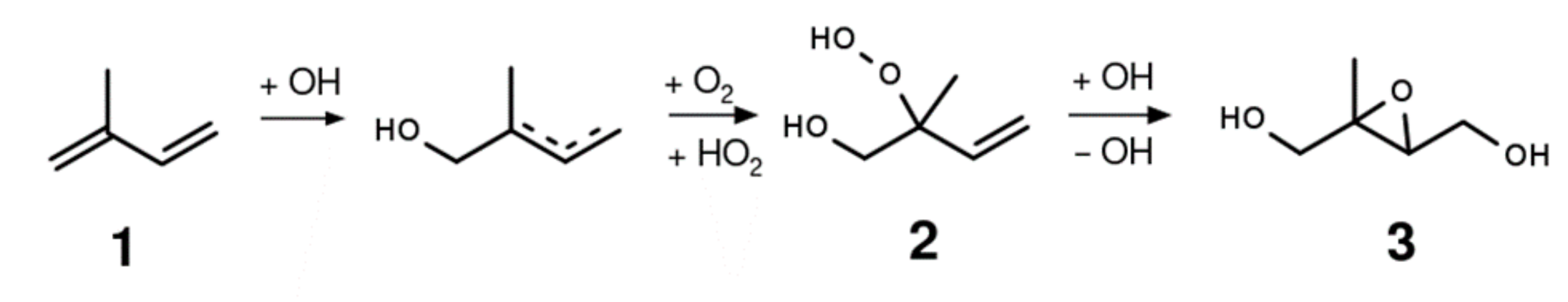
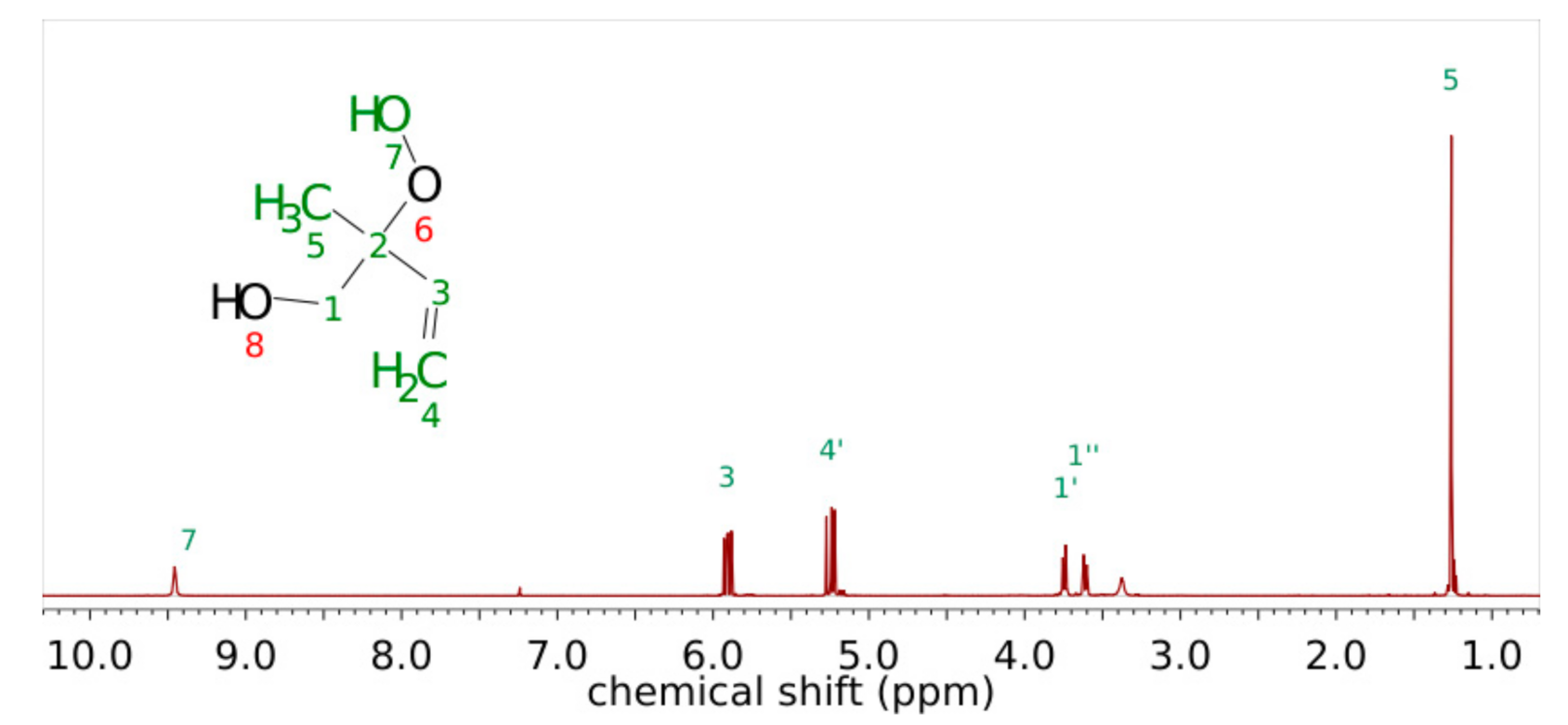
2.2. Synthesis of 1,2-ISOPOOH
2.3. Synthesis of 4,3-ISOPOOH
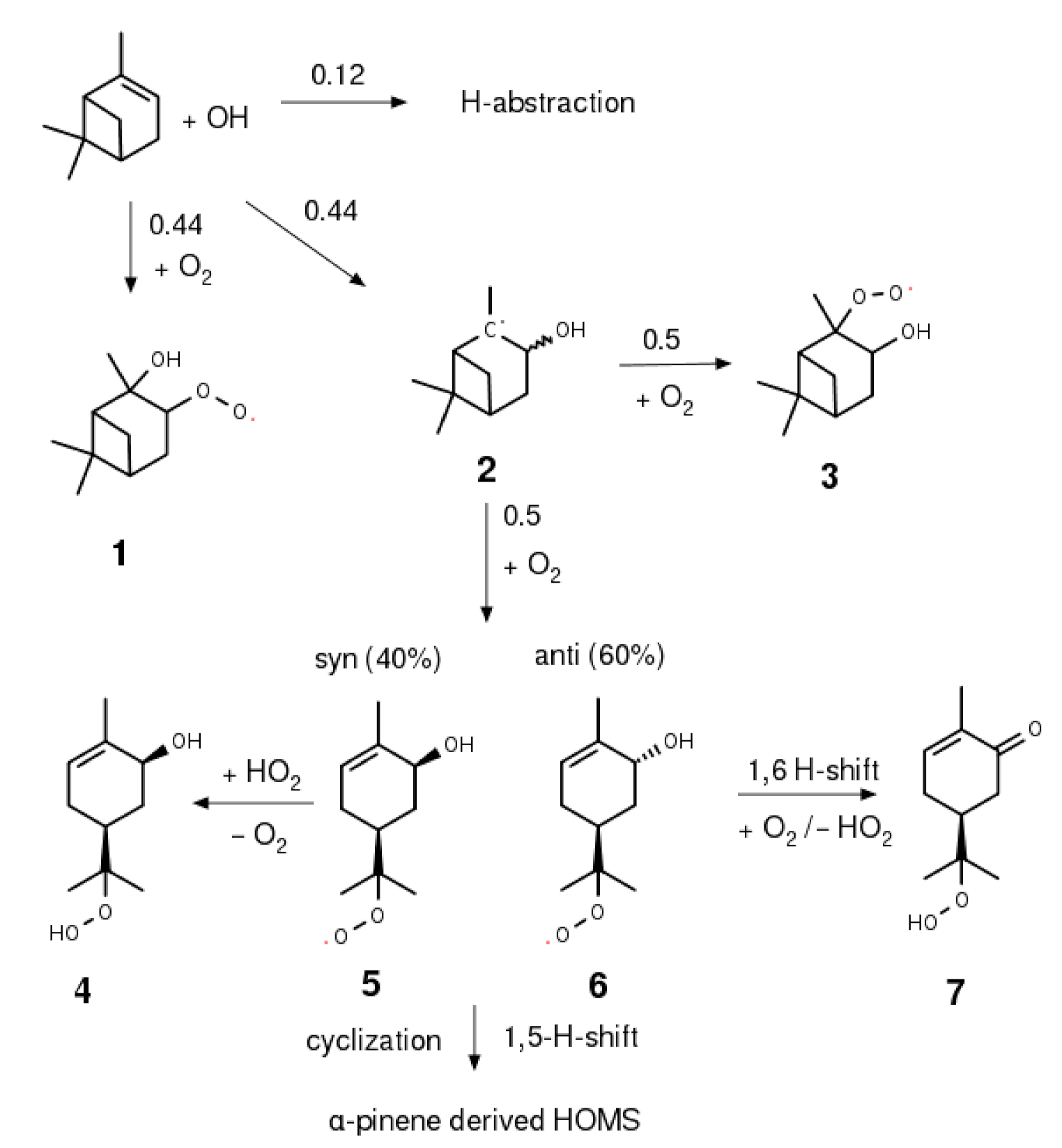
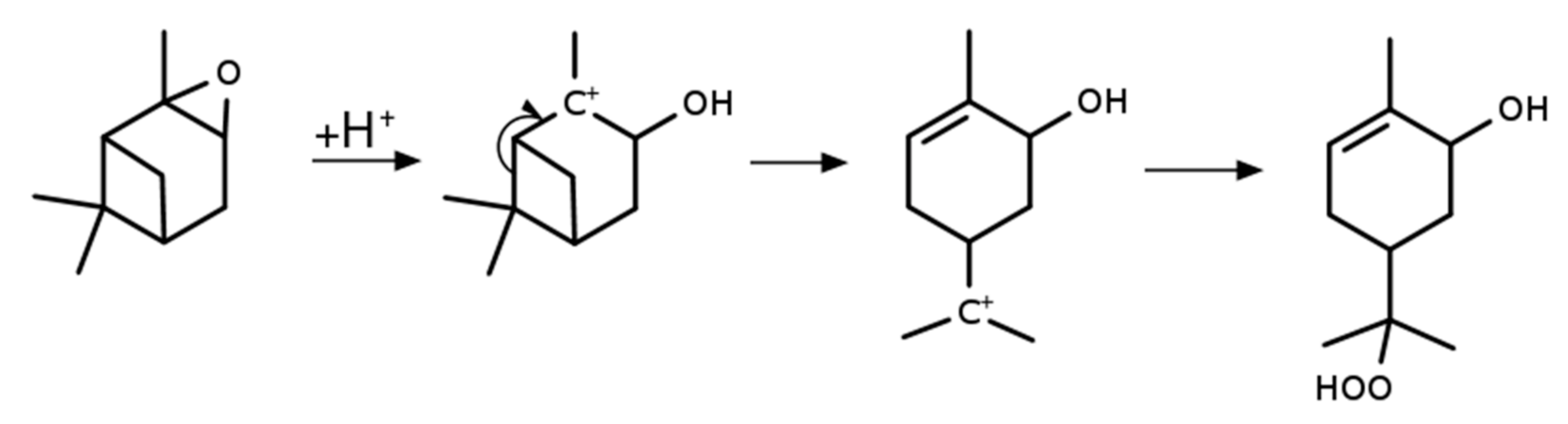
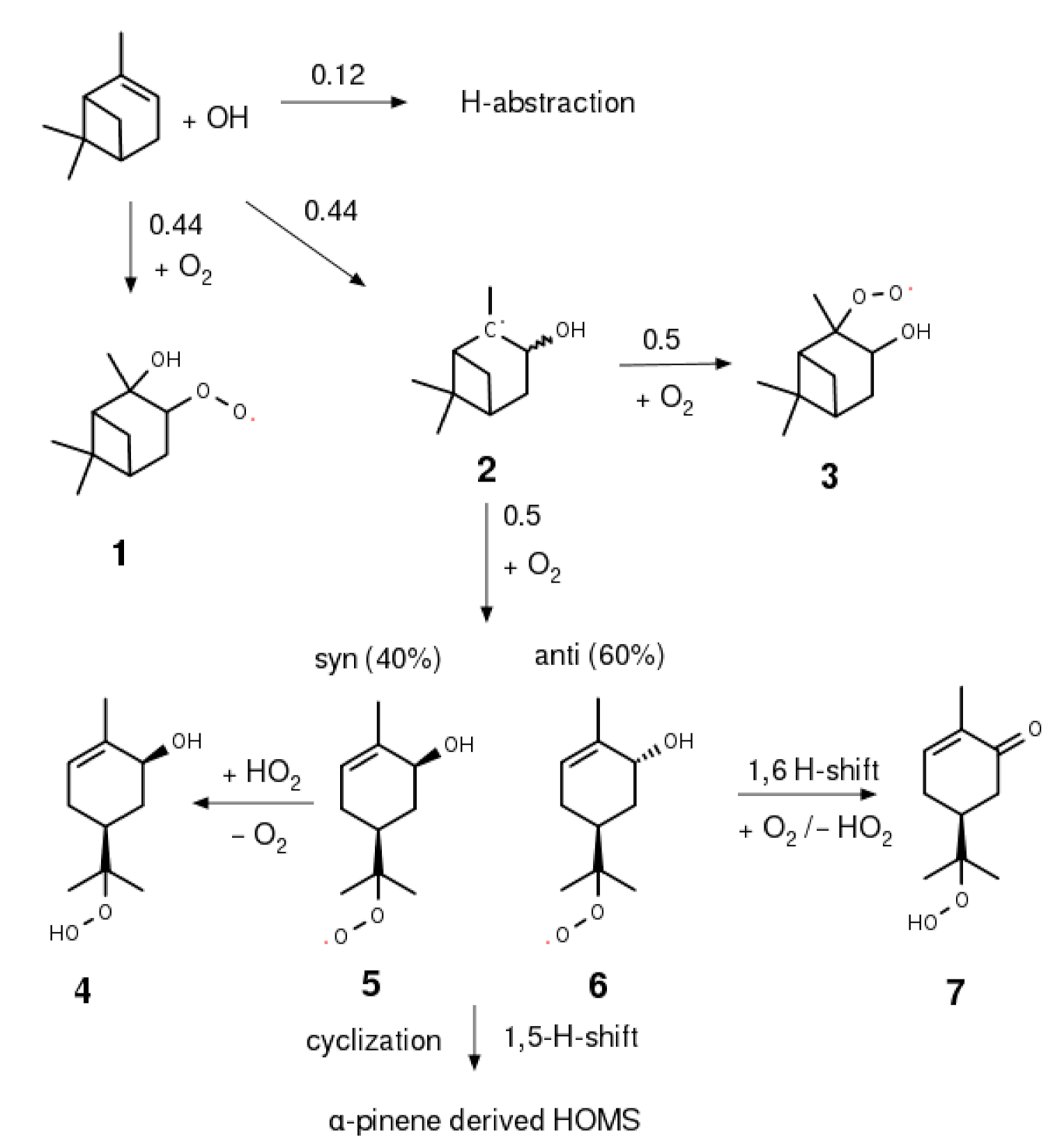
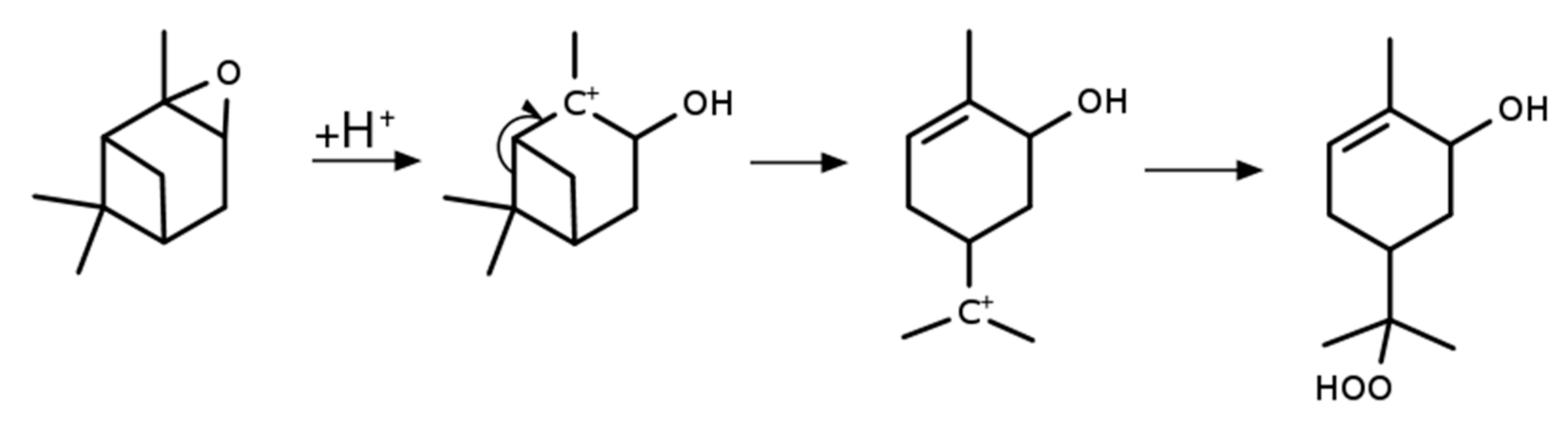
2.4. Synthesis of α-Pinene Hydroxy Hydroperoxide
2.5. ACD-C Aerosol-Chamber Setup
3. Results and Discussion
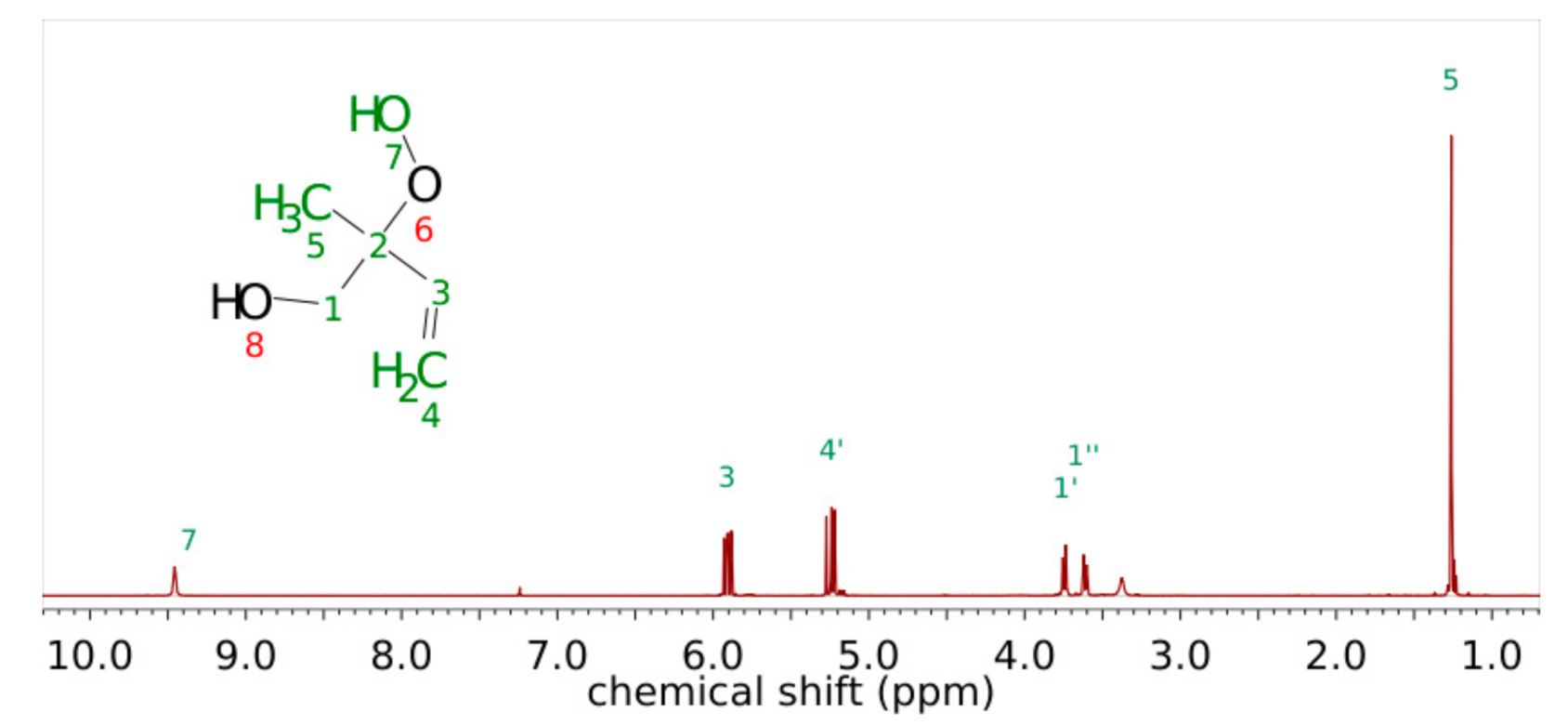
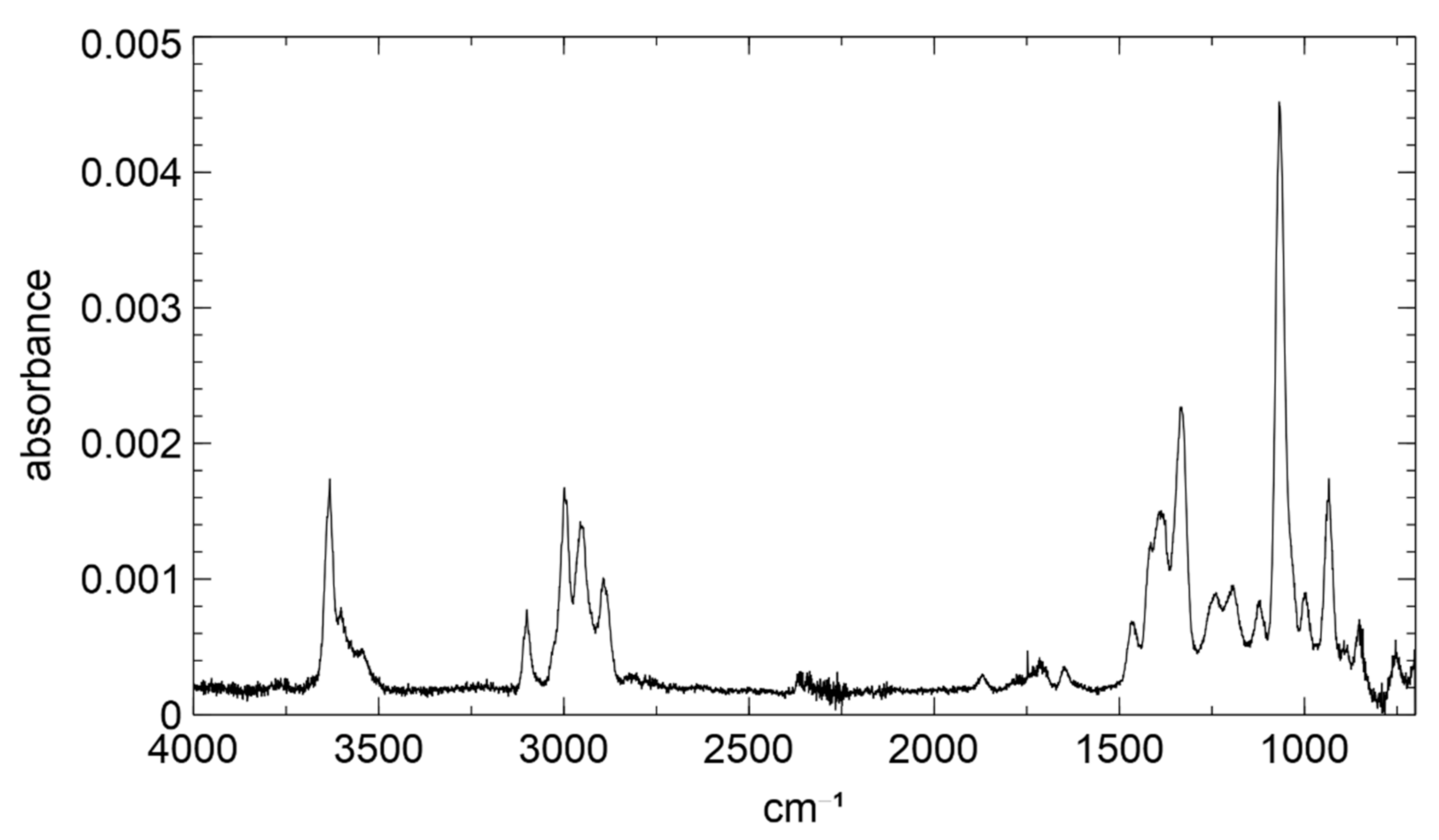
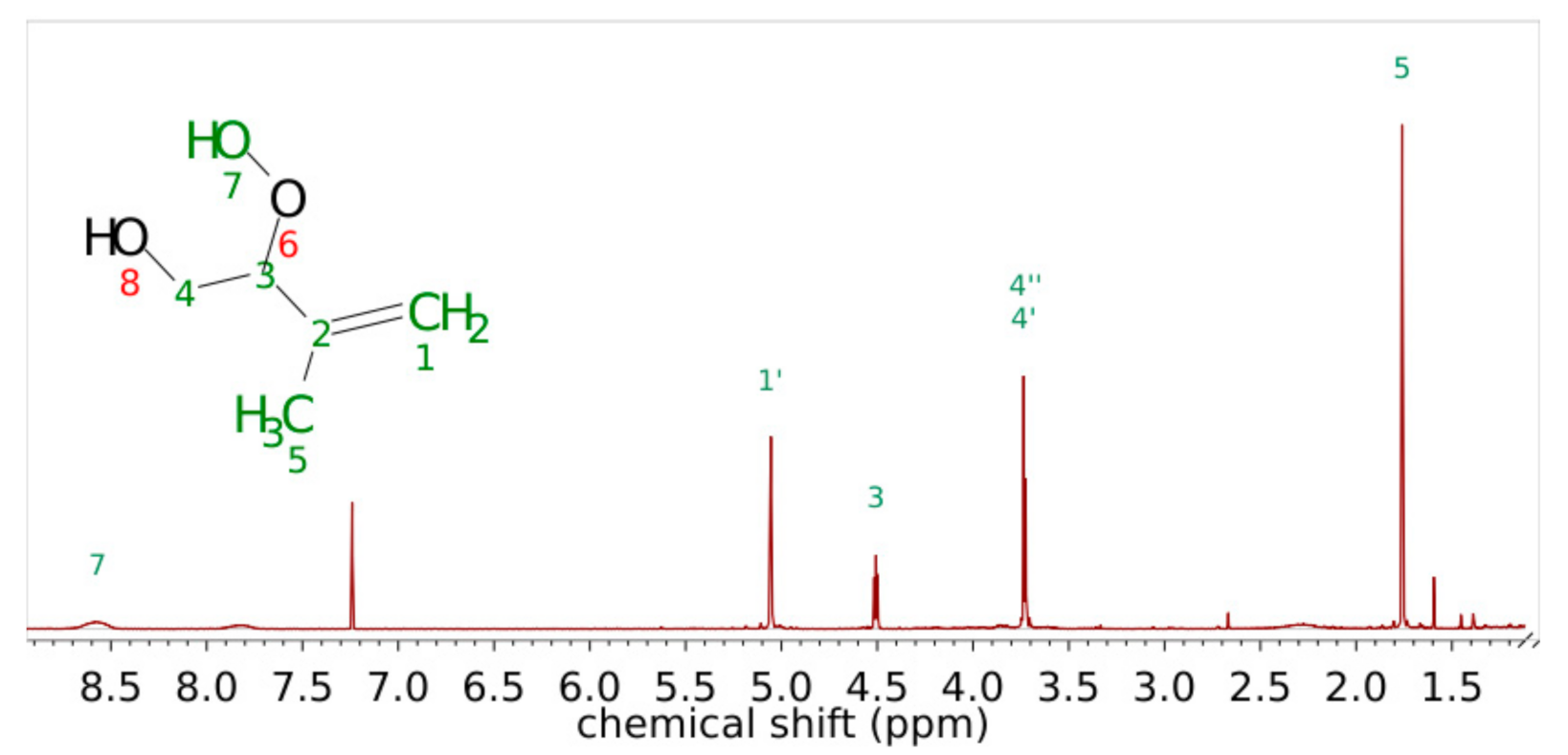
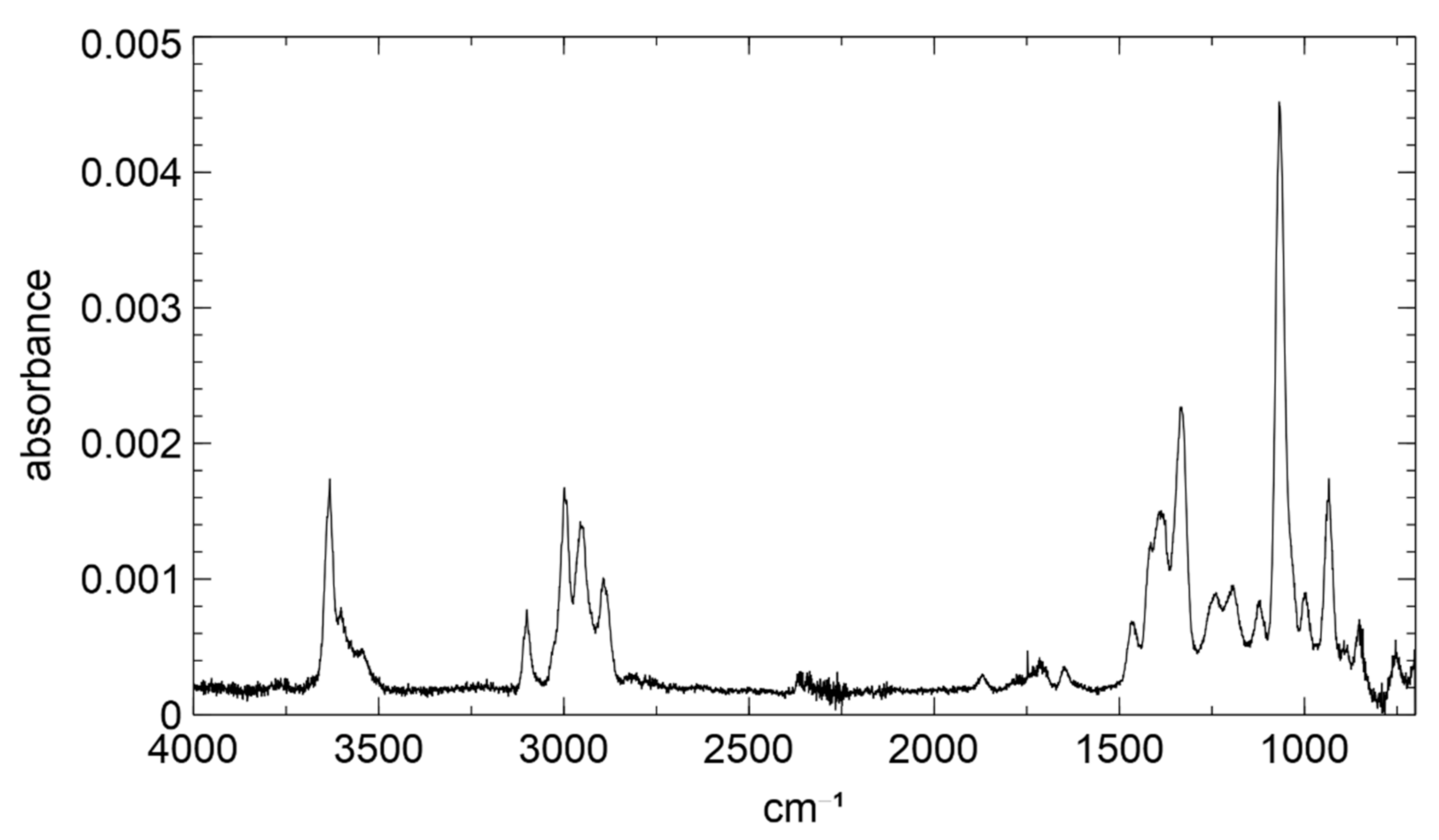
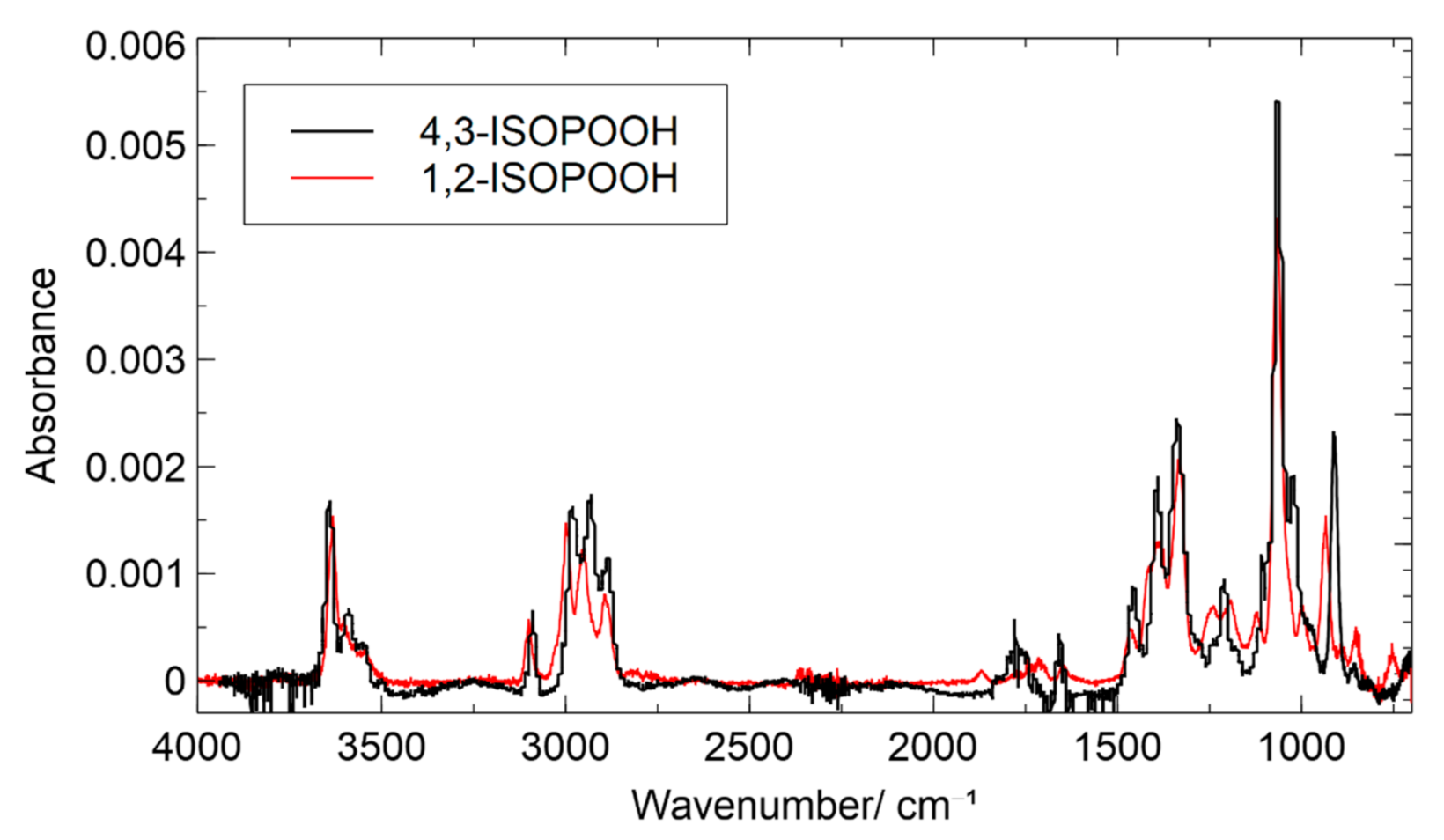
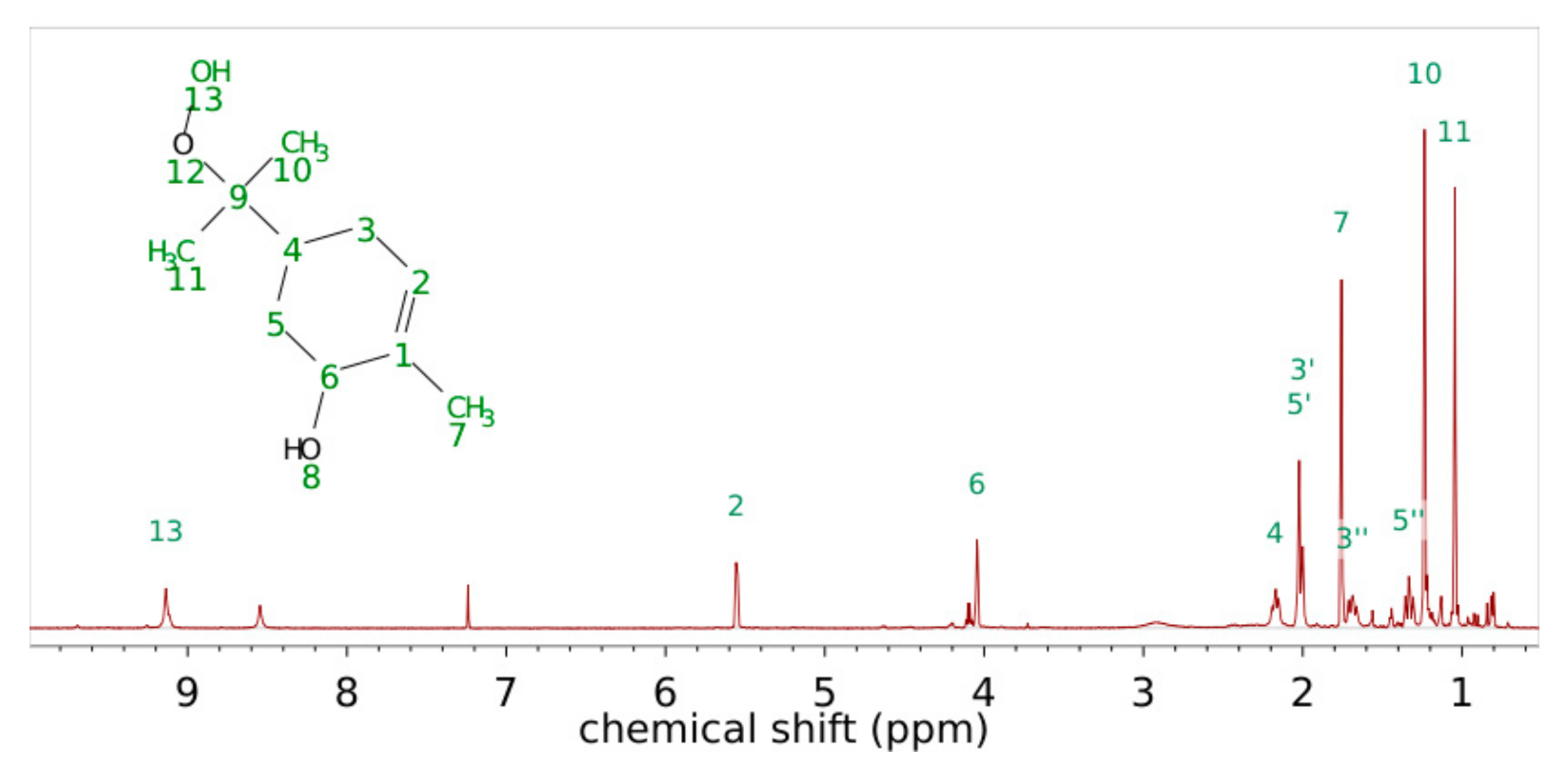
3.1. Product Characterization following the 1,2-ISOPOOH Synthesis
3.2. Product Characterization following the 4,3-ISOPOOH Synthesis
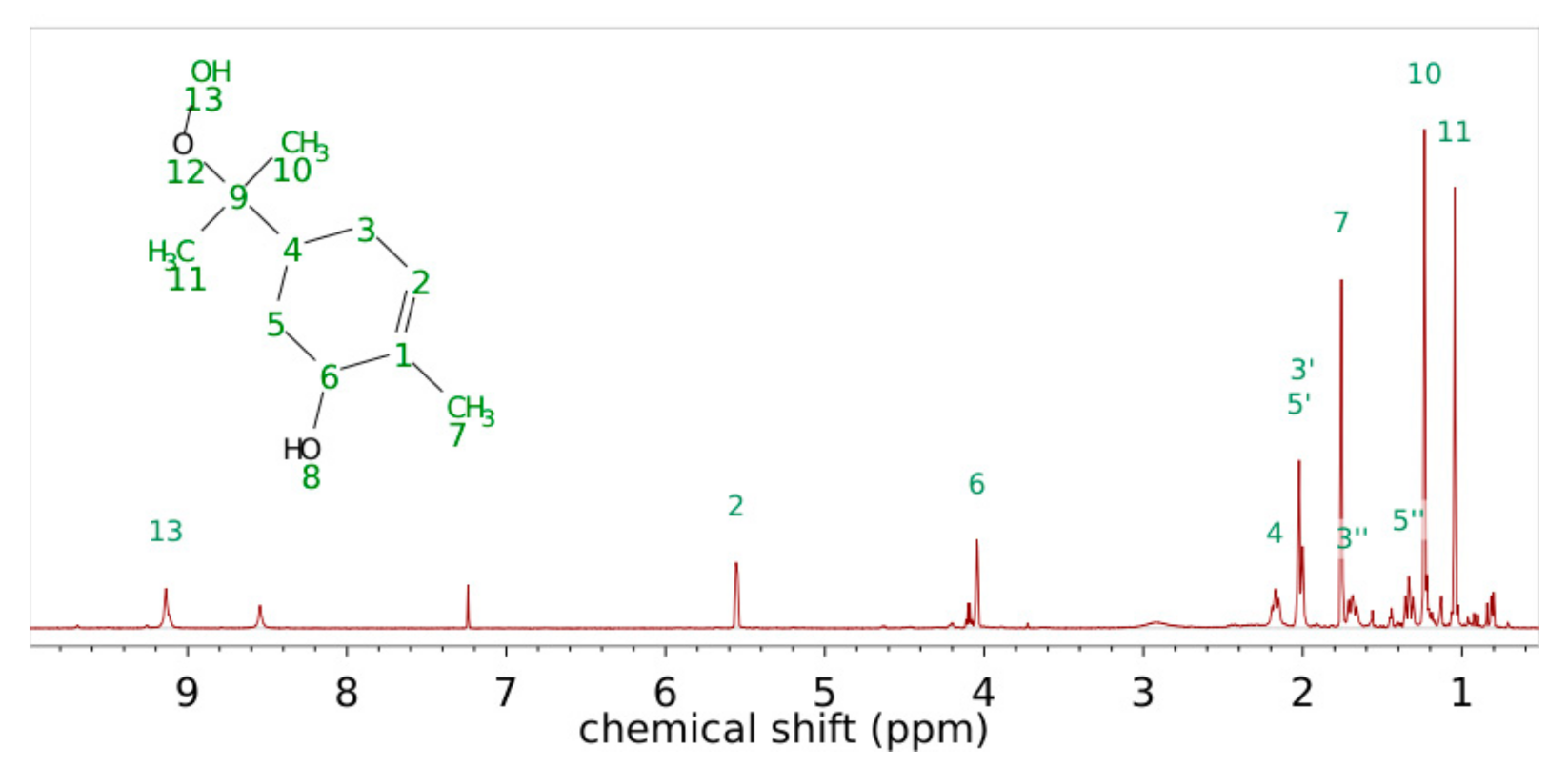
3.3. α-Pinene Hydroxy Hydroperoxide Synthesis
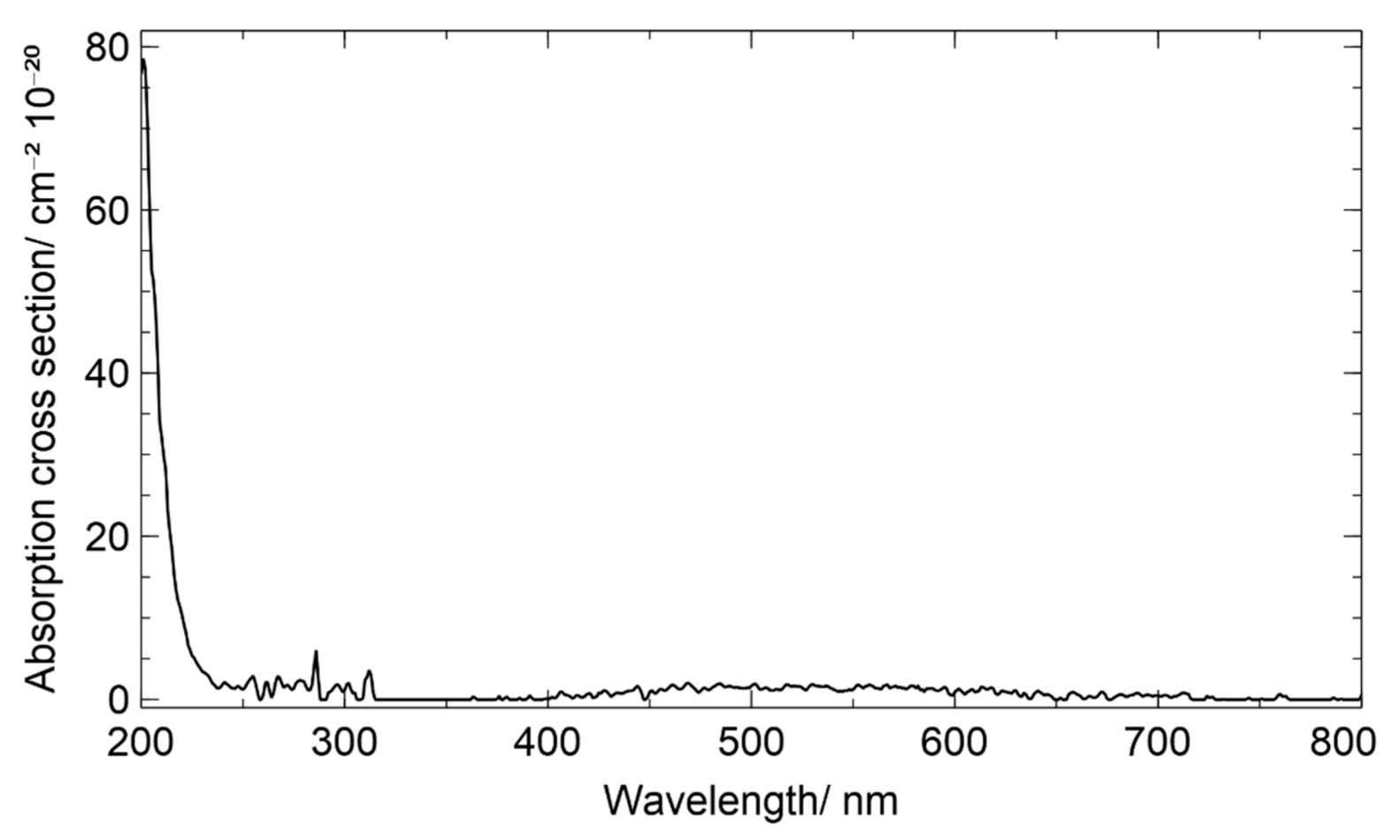
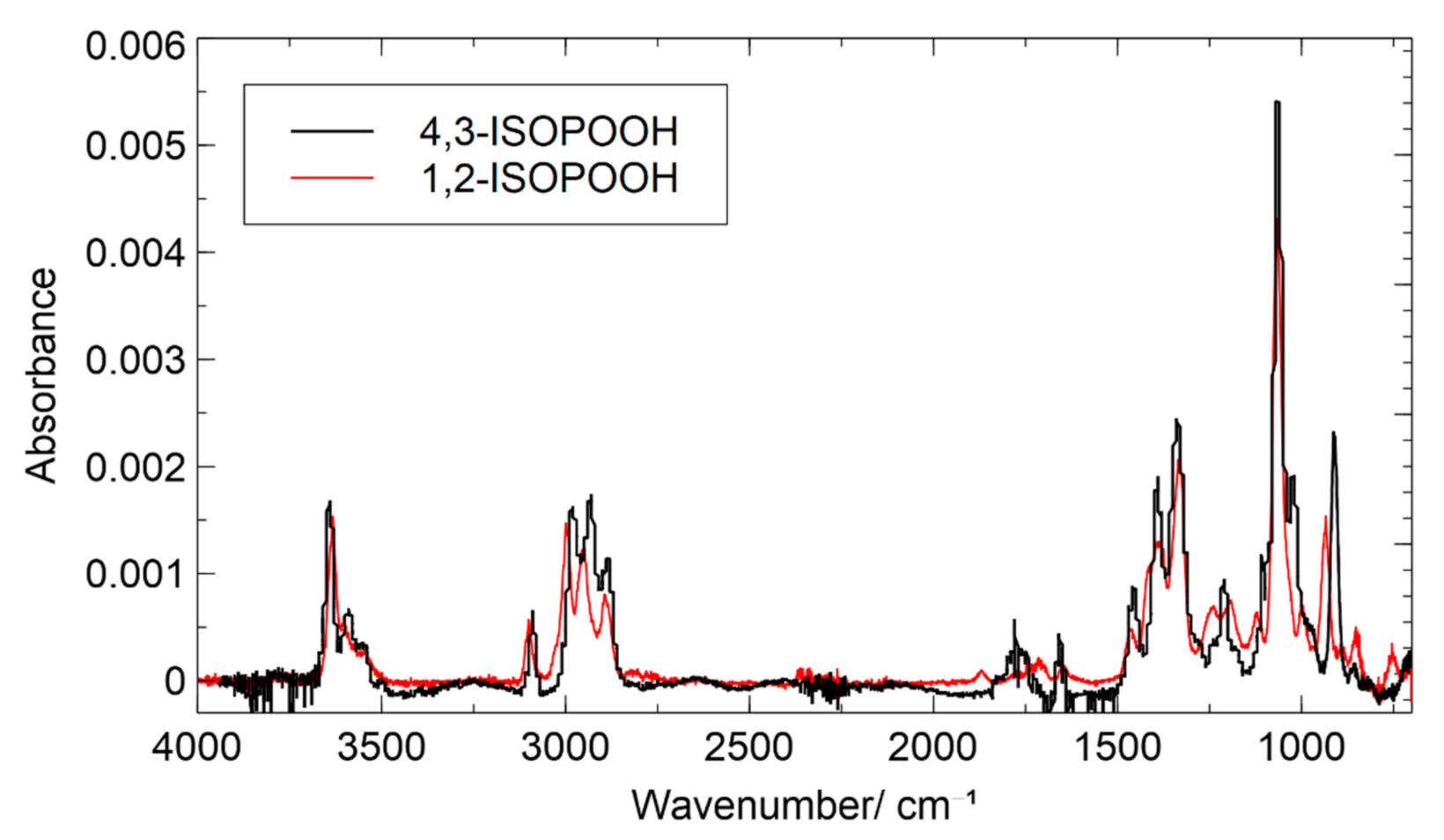
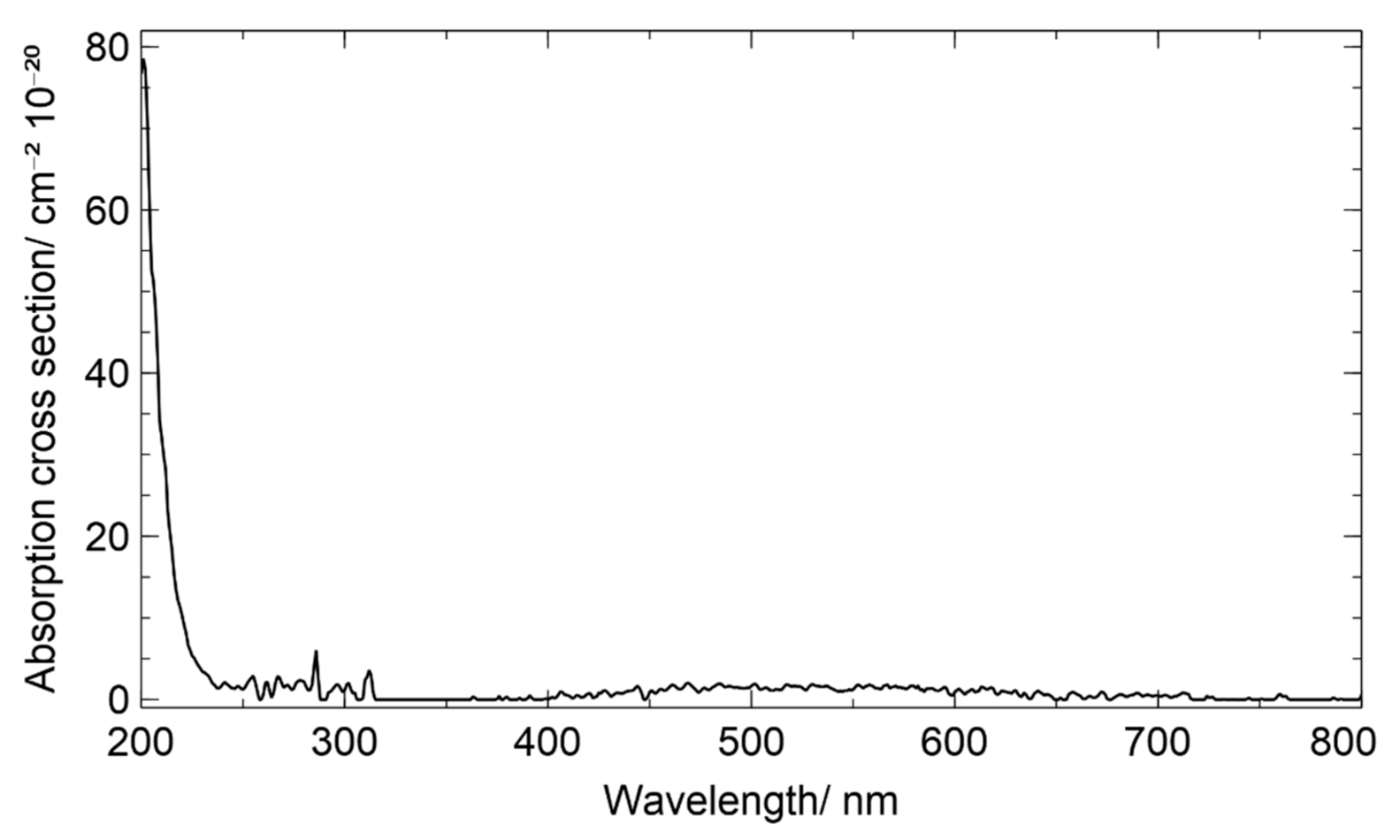
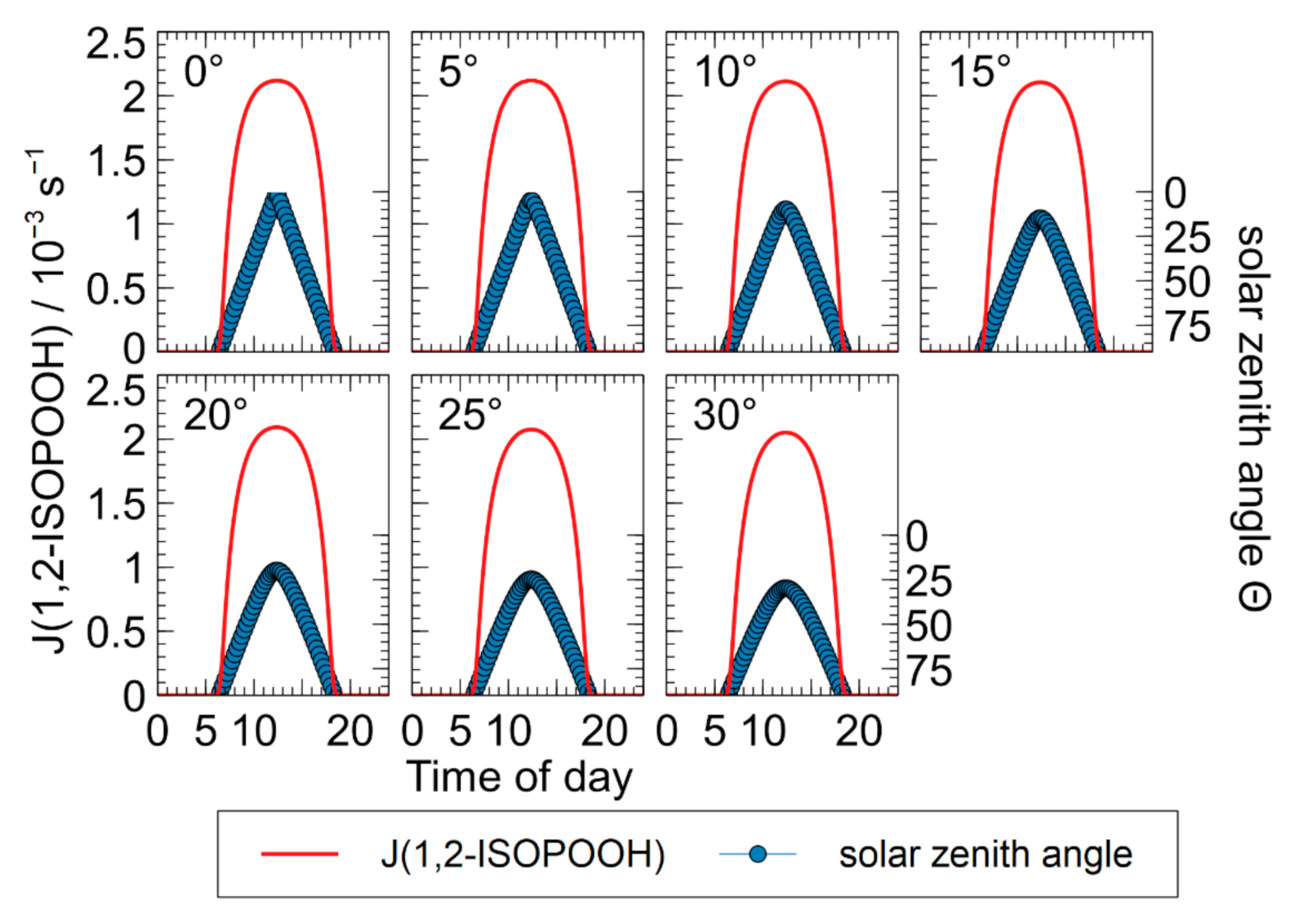
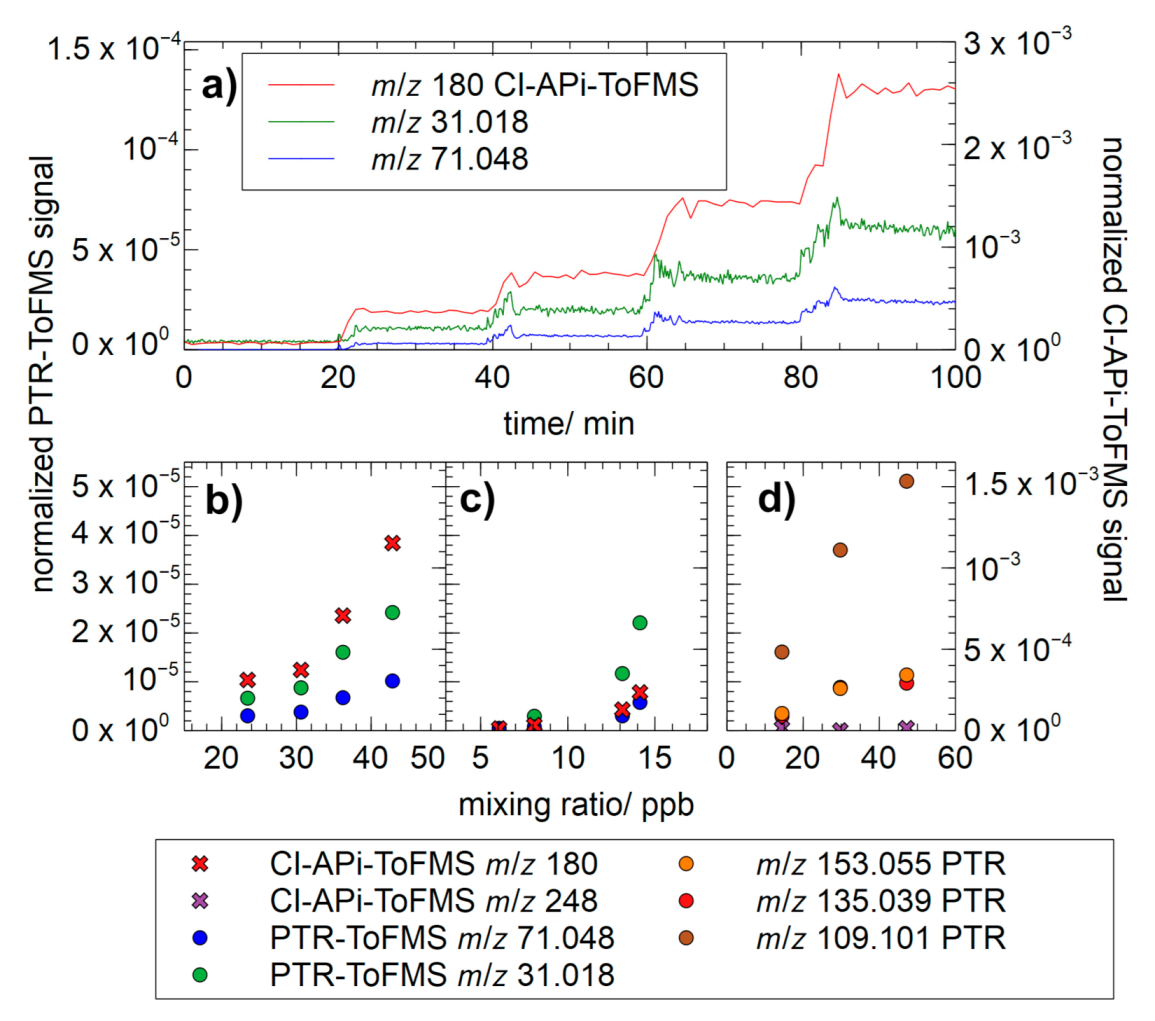
3.4. Chamber Characterization of Hydroxy Hydroperoxides
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef] [Green Version]
- Sindelarova, K.; Granier, C.; Bouarar, I.; Guenther, A.; Tilmes, S.; Stavrakou, T.; Müller, J.F.; Kuhn, U.; Stefani, P.; Knorr, W. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years. Atmos. Chem. Phys. 2014, 14, 9317–9341. [Google Scholar] [CrossRef] [Green Version]
- Patra, P.K.; Krol, M.C.; Prinn, R.G.; Takigawa, M.; Mühle, J.; Montzka, S.A.; Lal, S.; Yamashita, Y.; Naus, S.; Chandra, N.; et al. Methyl Chloroform Continues to Constrain the Hydroxyl (OH) Variability in the Troposphere. J. Geophys. Res. Atmos. 2021, 126, 1–15. [Google Scholar] [CrossRef]
- Matsumi, Y.; Kawasaki, M. Photolysis of Atmospheric Ozone in the Ultraviolet Region. Chem. Rev. 2003, 103, 4767–4782. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, J.; Butler, T.M.; Crowley, J.N.; Dillon, T.J.; Fischer, H.; Ganzeveld, L.; Harder, H.; Lawrence, M.G.; Martinez, M.; Taraborrelli, D.; et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature 2008, 452, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Barker, J.R.; Song, K. OH + Isoprene: A Direct Dynamics Study. Bull. Korean Chem. Soc. 2017, 38, 651–660. [Google Scholar] [CrossRef]
- Teng, A.P.; Crounse, J.D.; Wennberg, P.O. Isoprene Peroxy Radical Dynamics. J. Am. Chem. Soc. 2017, 139, 5367–5377. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.A.; Mills, J.H.; Porter, N.A. Theoretical Calculations of Carbon−Oxygen Bond Dissociation Enthalpies of Peroxyl Radicals Formed in the Autoxidation of Lipids. J. Am. Chem. Soc. 2003, 125, 5801–5810. [Google Scholar] [CrossRef]
- Peeters, J.; Nguyen, T.L.; Vereecken, L. HOx radical regeneration in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2009, 11, 5935–5939. [Google Scholar] [CrossRef]
- Crounse, J.D.; Paulot, F.; Kjaergaard, H.G.; Wennberg, P.O. Peroxy radical isomerization in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2011, 13, 13607–13613. [Google Scholar] [CrossRef] [Green Version]
- Berndt, T.; Hyttinen, N.; Herrmann, H.; Hansel, A. First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions. Commun. Chem. 2019, 2, 21. [Google Scholar] [CrossRef]
- D’Ambro, E.L.; Moller, K.H.; Lopez-Hilfiker, F.D.; Schobesberger, S.; Liu, J.; Shilling, J.E.; Lee, B.H.; Kjaergaard, H.G.; Thornton, J.A. Isomerization of Second-Generation Isoprene Peroxy Radicals: Epoxide Formation and Implications for Secondary Organic Aerosol Yields. Env. Sci. Technol. 2017, 51, 4978–4987. [Google Scholar] [CrossRef]
- Jenkin, M.E.; Boyd, A.A.; Lesclaux, R. Peroxy Radical Kinetics Resulting from the OH-Initiated Oxidation of 1,3-Butadiene, 2,3-Dimethyl-1,3-Butadiene and Isoprene. J. Atmos. Chem. 1998, 29, 267–298. [Google Scholar] [CrossRef]
- Tyndall, G.S.; Cox, R.A.; Granier, C.; Lesclaux, R.; Moortgat, G.K.; Pilling, M.J.; Ravishankara, A.R.; Wallington, T.J. Atmospheric chemistry of small organic peroxy radicals. J. Geophys. Res. Atmos. 2001, 106, 12157–12182. [Google Scholar] [CrossRef]
- Orlando, J.J.; Tyndall, G.S. Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev. 2012, 41, 6294–6317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, T.; Scholz, W.; Mentler, B.; Fischer, L.; Herrmann, H.; Kulmala, M.; Hansel, A. Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere. Angew. Chem. Int. Ed. 2018, 57, 3820–3824. [Google Scholar] [CrossRef]
- Wang, Y.; Mehra, A.; Krechmer, J.E.; Yang, G.; Hu, X.; Lu, Y.; Lambe, A.; Canagaratna, M.; Chen, J.; Worsnop, D.; et al. Oxygenated products formed from OH-initiated reactions of trimethylbenzene: Autoxidation and accretion. Atmos. Chem. Phys. 2020, 20, 9563–9579. [Google Scholar] [CrossRef]
- Tomaz, S.; Wang, D.; Zabalegui, N.; Li, D.; Lamkaddam, H.; Bachmeier, F.; Vogel, A.; Monge, M.E.; Perrier, S.; Baltensperger, U.; et al. Structures and reactivity of peroxy radicals and dimeric products revealed by online tandem mass spectrometry. Nat. Commun. 2021, 12, 300. [Google Scholar] [CrossRef]
- Vaughan, S.; Ingham, T.; Whalley, L.K.; Stone, D.; Evans, M.J.; Read, K.A.; Lee, J.D.; Moller, S.J.; Carpenter, L.J.; Lewis, A.C.; et al. Seasonal observations of OH and HO<sub>2</sub> in the remote tropical marine boundary layer. Atmos. Chem. Phys. 2012, 12, 2149–2172. [Google Scholar] [CrossRef] [Green Version]
- Madronich, S.; Calvert, J.G. Permutation reactions of organic peroxy radicals in the troposphere. J. Geophys. Res. Atmos. 1990, 95, 5697–5715. [Google Scholar] [CrossRef]
- Holzinger, R.; Sanhueza, E.; von Kuhlmann, R.; Kleiss, B.; Donoso, L.; Crutzen, P.J. Diurnal cycles and seasonal variation of isoprene and its oxidation products in the tropical savanna atmosphere. Glob. Biogeochem. Cycles 2002, 16, 22-1–22-13. [Google Scholar] [CrossRef]
- Crutzen, P.J.; Williams, J.; Pöschl, U.; Hoor, P.; Fischer, H.; Warneke, C.; Holzinger, R.; Hansel, A.; Lindinger, W.; Scheeren, B.; et al. High spatial and temporal resolution measurements of primary organics and their oxidation products over the tropical forests of Surinam. Atmos. Environ. 2000, 34, 1161–1165. [Google Scholar] [CrossRef]
- Paulot, F.; Crounse, J.D.; Kjaergaard, H.G.; Kürten, A.; St. Clair, J.M.; Seinfeld, J.H.; Wennberg, P.O. Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene. Science 2009, 325, 730. [Google Scholar] [CrossRef] [Green Version]
- Crounse, J.D.; McKinney, K.A.; Kwan, A.J.; Wennberg, P.O. Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry. Anal. Chem. 2006, 78, 6726–6732. [Google Scholar] [CrossRef] [Green Version]
- Vereecken, L.; Peeters, J. Theoretical Study of the Formation of Acetone in the OH-Initiated Atmospheric Oxidation of α-Pinene. J. Phys. Chem. A 2000, 104, 11140–11146. [Google Scholar] [CrossRef]
- Berndt, T.; Richters, S.; Jokinen, T.; Hyttinen, N.; Kurten, T.; Otkjaer, R.V.; Kjaergaard, H.G.; Stratmann, F.; Herrmann, H.; Sipila, M.; et al. Hydroxyl radical-induced formation of highly oxidized organic compounds. Nat. Commun. 2016, 7, 13677. [Google Scholar] [CrossRef] [PubMed]
- Vereecken, L.; Muller, J.F.; Peeters, J. Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of alpha-pinene: Impact of non-traditional peroxyl radical chemistry. Phys. Chem. Chem. Phys. 2007, 9, 5241–5248. [Google Scholar] [CrossRef] [PubMed]
- Peeters, J.; Vereecken, L.; Fantechi, G. The detailed mechanism of the OH-initiated atmospheric oxidation of α-pinene: A theoretical study. Phys. Chem. Chem. Phys. 2001, 3, 5489–5504. [Google Scholar] [CrossRef]
- Berndt, T. Peroxy Radical Processes and Product Formation in the OH Radical-Initiated Oxidation of alpha-Pinene for Near-Atmospheric Conditions. J. Phys. Chem. A 2021, 125, 9151–9160. [Google Scholar] [CrossRef]
- Xu, L.; Moller, K.H.; Crounse, J.D.; Otkjwr, R.V.; Kjaergaard, H.G.; Wennberg, P.O. Unimolecular Reactions of Peroxy Radicals Formed in the Oxidation of alpha-Pinene and beta-Pinene by Hydroxyl Radicals. J. Phys. Chem. A 2019, 123, 1661–1674. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Tsona, N.T.; Du, L. Relative Humidity Changes the Role of SO2 in Biogenic Secondary Organic Aerosol Formation. J. Phys. Chem. Lett. 2021, 12, 7365–7372. [Google Scholar] [CrossRef]
- Madronich, S.; Flocke, S. Theoretical Estimation of Biologically Effective UV Radiation at the Earth’s Surface. In Solar Ultraviolet Radiation, Proceedings of NATO ASI Series, Halkidiki, Greece, 2–11 October 1995; Zerefos, C.S., Bais, A.F., Eds.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 23–48. [Google Scholar]
- Saunders, S.M.; Jenkin, M.E.; Derwent, R.G.; Pilling, M.J. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.W.; LaCount, R.B. The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides1. J. Am. Chem. Soc. 1961, 83, 417–423. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Mutzel, A.; Poulain, L.; Berndt, T.; Iinuma, Y.; Rodigast, M.; Böge, O.; Richters, S.; Spindler, G.; Sipila, M.; Jokinen, T.; et al. Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study. Environ. Sci. Technol. 2015, 49, 7754–7761. [Google Scholar] [CrossRef]
- Junninen, H.; Ehn, M.; Petäjä, T.; Luosujärvi, L.; Kotiaho, T.; Kostiainen, R.; Rohner, U.; Gonin, M.; Fuhrer, K.; Kulmala, M.; et al. A high-resolution mass spectrometer to measure atmospheric ion composition. Atmos. Meas. Tech. 2010, 3, 1039–1053. [Google Scholar] [CrossRef] [Green Version]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Processes 1998, 173, 191–241. [Google Scholar] [CrossRef]
- Birmili, W.; Stratmann, F.; Wiedensohler, A. Design of a Dma-Based Size Spectrometer for a Large Particle Size Range and Stable Operation. J. Aerosol Sci. 1999, 30, 549–553. [Google Scholar] [CrossRef]
- Riva, M.; Budisulistiorini, S.H.; Chen, Y.; Zhang, Z.; D’Ambro, E.L.; Zhang, X.; Gold, A.; Turpin, B.J.; Thornton, J.A.; Canagaratna, M.R.; et al. Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides. Env. Sci. Technol. 2016, 50, 9889–9899. [Google Scholar] [CrossRef]
- Li, Y.; Hao, H.-D.; Wu, Y. Facile Ring-Opening of Oxiranes by H2O2 Catalyzed by Phosphomolybdic Acid. Org. Lett. 2009, 11, 2691–2694. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Zhang, Z.-H.; Li, T.-S. Efficient Conversion of Epoxides into β-Hydroperoxy Alcohols Catalyzed by Antimony Trichloride/SiO2. Synthesis 2008, 2008, 3314–3318. [Google Scholar] [CrossRef]
- Budisulistiorini, S.H.; Li, X.; Bairai, S.T.; Renfro, J.; Liu, Y.; Liu, Y.J.; McKinney, K.A.; Martin, S.T.; McNeill, V.F.; Pye, H.O.T.; et al. Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site. Atmos. Chem. Phys. 2015, 15, 8871–8888. [Google Scholar] [CrossRef] [Green Version]
- Pires, R.V.; Pessoa, L.M.B.; Sant’Anna, M.d.A.d.; Fainleib, A.; Nunes, R.d.C.P.; Lucas, E.F. Synthesis and characterization of isoprene oligomers to compare different production chemical processes. Polímeros 2019, 29, 1–9. [Google Scholar] [CrossRef]
- Dovrou, E.; Bates, K.H.; Rivera-Rios, J.C.; Cox, J.L.; Shutter, J.D.; Keutsch, F.N. Towards a chemical mechanism of the oxidation of aqueous sulfur dioxide via isoprene hydroxyl hydroperoxides (ISOPOOH). Atmos. Chem. Phys. 2021, 21, 8999–9008. [Google Scholar] [CrossRef]
- Zanca, N.; Lambe, A.T.; Massoli, P.; Paglione, M.; Croasdale, D.R.; Parmar, Y.; Tagliavini, E.; Gilardoni, S.; Decesari, S. Characterizing source fingerprints and ageing processes in laboratory-generated secondary organic aerosols using proton-nuclear magnetic resonance (1H-NMR) analysis and HPLC HULIS determination. Atmos. Chem. Phys. 2017, 17, 10405–10421. [Google Scholar] [CrossRef] [Green Version]
- Bates, K.H.; Crounse, J.D.; St Clair, J.M.; Bennett, N.B.; Nguyen, T.B.; Seinfeld, J.H.; Stoltz, B.M.; Wennberg, P.O. Gas phase production and loss of isoprene epoxydiols. J. Phys. Chem. A 2014, 118, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Shreve, O.D.; Heether, M.R.; Knight, H.B.; Swern, D. Infrared Absorption Spectra of Some Hydroperoxides, Peroxides, and Related Compounds. Anal. Chem. 1951, 23, 282–285. [Google Scholar] [CrossRef]
- Brauer, C.S.; Blake, T.A.; Guenther, A.B.; Sharpe, S.W.; Sams, R.L.; Johnson, T.J. Quantitative infrared absorption cross sections of isoprene for atmospheric measurements. Atmos. Meas. Tech. 2014, 7, 3839–3847. [Google Scholar] [CrossRef] [Green Version]
- Bernhammer, A.-K.; Breitenlechner, M.; Keutsch, F.N.; Hansel, A. Technical note: Conversion of isoprene hydroxy hydroperoxides (ISOPOOHs) on metal environmental simulation chamber walls. Atmos. Chem. Phys. 2017, 17, 4053–4062. [Google Scholar] [CrossRef] [Green Version]
- Vacque, V.; Sombret, B.; Huvenne, J.P.; Legrand, P.; Suc, S. Characterisation of the O-O peroxide bond by vibrational spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 55–66. [Google Scholar] [CrossRef]
- Mohnhaupt, M.; Hagemann, H.; Perler, J.-P.; Bill, H.; Boukouvalas, J.; Rossier, J.-C.; Jefford, C.W. A Vibrational Study of Some 1,2,4-Trioxanes. Helv. Chim. Acta 1988, 71, 992–999. [Google Scholar] [CrossRef]
- Vaghjiani, G.L.; Ravishankara, A.R. Absorption cross sections of CH3OOH, H2O2, and D2O2 vapors between 210 and 365 nm at 297 K. J. Geophys. Res. Atmos. 1989, 94, 3487–3492. [Google Scholar] [CrossRef]
- Matthews, J.; Sinha, A.; Francisco, J.S. The importance of weak absorption features in promoting tropospheric radical production. Proc. Natl. Acad. Sci. USA 2005, 102, 7449. [Google Scholar] [CrossRef] [Green Version]
- Wennberg, P.O.; Bates, K.H.; Crounse, J.D.; Dodson, L.G.; McVay, R.C.; Mertens, L.A.; Nguyen, T.B.; Praske, E.; Schwantes, R.H.; Smarte, M.D.; et al. Gas-Phase Reactions of Isoprene and Its Major Oxidation Products. Chem. Rev. 2018, 118, 3337–3390. [Google Scholar] [CrossRef] [Green Version]
- St Clair, J.M.; Rivera-Rios, J.C.; Crounse, J.D.; Knap, H.C.; Bates, K.H.; Teng, A.P.; Jorgensen, S.; Kjaergaard, H.G.; Keutsch, F.N.; Wennberg, P.O. Kinetics and Products of the Reaction of the First-Generation Isoprene Hydroxy Hydroperoxide (ISOPOOH) with OH. J. Phys. Chem. A 2016, 120, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Jenkin, M.E.; Saunders, S.M.; Pilling, M.J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development. Atmos. Environ. 1997, 31, 81–104. [Google Scholar] [CrossRef]
- Forrester, J.; Jones, R.V.H.; Preston, P.N.; Simpson, E.S.C. Efficient use of trimethylsulfonium methylsulfate as a reagent for the epoxidation of carbonyl-containing compounds. J. Chem. Soc. Perkin Trans. 1999, 22, 3333–3335. [Google Scholar] [CrossRef]
- Ehn, M.; Thornton, J.A.; Kleist, E.; Sipilä, M.; Junninen, H.; Pullinen, I.; Springer, M.; Rubach, F.; Tillmann, R.; Lee, B.; et al. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–479. [Google Scholar] [CrossRef]
- Kürten, A.; Rondo, L.; Ehrhart, S.; Curtius, J. Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid. J. Phys. Chem. A 2012, 116, 6375–6386. [Google Scholar] [CrossRef]
- Bianchi, F.; Kurtén, T.; Riva, M.; Mohr, C.; Rissanen, M.P.; Roldin, P.; Berndt, T.; Crounse, J.D.; Wennberg, P.O.; Mentel, T.F.; et al. Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol. Chem. Rev. 2019, 119, 3472–3509. [Google Scholar] [CrossRef] [Green Version]
- Vasquez, K.T.; Allen, H.M.; Crounse, J.D.; Praske, E.; Xu, L.; Noelscher, A.C.; Wennberg, P.O. Low-pressure gas chromatography with chemical ionization mass spectrometry for quantification of multifunctional organic compounds in the atmosphere. Atmos. Meas. Tech. 2018, 11, 6815–6832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Rivera-Rios, J.C.; Keutsch, F.N.; Abbatt, J.P.D. Identification of organic hydroperoxides and peroxy acids using atmospheric pressure chemical ionization–tandem mass spectrometry (APCI-MS/MS): Application to secondary organic aerosol. Atmos. Meas. Tech. 2018, 11, 3081–3089. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Rios, J.C.; Nguyen, T.B.; Crounse, J.D.; Jud, W.; St. Clair, J.M.; Mikoviny, T.; Gilman, J.B.; Lerner, B.M.; Kaiser, J.B.; de Gouw, J.; et al. Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry. Geophys. Res. Lett. 2014, 41, 8645–8651. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; Herdlinger-Blatt, I.; McKinney, K.A.; Martin, S.T. Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation. Atmos. Chem. Phys. 2013, 13, 5715–5730. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Lakey, P.S.J.; Rivera-Rios, J.C.; Keutsch, F.N.; Shiraiwa, M. Aqueous-Phase Decomposition of Isoprene Hydroxy Hydroperoxide and Hydroxyl Radical Formation by Fenton-like Reactions with Iron Ions. J. Phys. Chem. A 2020, 124, 5230–5236. [Google Scholar] [CrossRef]
- Dovrou, E.; Rivera-Rios, J.C.; Bates, K.H.; Keutsch, F.N. Sulfate Formation via Cloud Processing from Isoprene Hydroxyl Hydroperoxides (ISOPOOH). Environ. Sci. Technol. 2019, 53, 12476–12484. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Atom Number | Shift of Hydrogen Atom in 1H-NMR/ppm | Shift of Carbon Atom in 13C-NMR/ppm | H-H Coupling COSY Coupling Constants J in Brackets (from 1H) | C-H Coupling HMBC |
|---|---|---|---|---|
| 1 | 134.22 | C1-H6; C1-H5′; C1-H7 | ||
| 2 | 5.56 | 125.85 | H2-H7; H2-H3′ (3Hz) | C2-H6; C2-H7 |
| 3 | 3′ 2.00 3″ 1.69 | 27.60 | H3′-H3″H3′-H2 (3 Hz); H3′-H4 H3″-H3′ (14.4 Hz); H3″-H4 (14.4 Hz) | C3-H2; C3-H5’ |
| 4 | 2.17 | 33.49 | H4-H3’ (14.4 Hz); H4-H3″; H4-H5′; H4-H5″ (13.5 Hz) | C4-H3′; C4-H3″; C4-H5′; C4-H5″, C4-H10; C4-H11 |
| 5 | 5′ 2.02 5″ 1.33 | 31.97 | H5′-H4, H5’-H5″ (13.6 Hz) H5″-H4 (13.6 Hz); H5″-H5′ (13.6 Hz); H5″-H6 | |
| 6 | 4.04 | 69.03 | H6-H5″, H6-H7 | C6-H2; C6-H5′; C6-H7 |
| 7 | 1.76 | 21.00 | H7-H6; H7-H3′ | |
| 9 | 84.45 | C9-H10; C9-H11 | ||
| 10 | 1.23 | 22.91 | C10-H11 | |
| 11 | 1.04 | 20.37 | C11-H10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mettke, P.; Mutzel, A.; Böge, O.; Herrmann, H. Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides. Atmosphere 2022, 13, 507. https://doi.org/10.3390/atmos13040507
Mettke P, Mutzel A, Böge O, Herrmann H. Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides. Atmosphere. 2022; 13(4):507. https://doi.org/10.3390/atmos13040507
Chicago/Turabian StyleMettke, Peter, Anke Mutzel, Olaf Böge, and Hartmut Herrmann. 2022. "Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides" Atmosphere 13, no. 4: 507. https://doi.org/10.3390/atmos13040507
APA StyleMettke, P., Mutzel, A., Böge, O., & Herrmann, H. (2022). Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides. Atmosphere, 13(4), 507. https://doi.org/10.3390/atmos13040507