
Rate Constants and Branching Ratios for the Self-Reaction of Acetyl Peroxy (CH3C(O)O2•) and Its Reaction with CH3O2



Abstract
:1. Introduction
- its self-reaction2 CH3C(O)O2• → 2 CH3C(O)O• + O2
- its reaction with CH3O2•CH3C(O)O2• + CH3O2• → CH3O• + CH3C(O)O• + O2whereby (R2a) maintains the radical pool, while (R2b) is a termination reaction. Therefore, a reliable determination of the branching ratio is of importance.→ CH3C(O)OH + HCHO + O2
- its reaction with HO2CH3C(O)O2• + HO2• → CH3C(O)OOH + O2→ CH3C(O)OH + O3→ CH3C(O)O• + •OH + O2
2. Experimental
2.1. Experimental Setup
- pulsed 351 nm photolysis of acetaldehyde (CH3CHO)/Cl2/O2 mixtures:Cl2 + hν351 nm → 2 Cl•CH3CHO + Cl• → CH3CO• + HClCH3CO• + O2 (+ M) → CH3C(O)O2• (+ M)CH3CO• can also react with O2 through other pathways: it has been observed [39,40] that its reaction with O2 can also lead to low concentrations of HO2• and •OH, depending on the amount of internal energy of CH3CO•:CH3CO• + O2 → •CH2CO + HO2•/CH2C(O)O• + •OHCH3CO• might also decompose before reaction with O2:CH3CO → CH3 + CO
- pulsed 351 nm photolysis of biacetyl (CH3C(O)C(O)CH3)/O2 mixtures:followed by either (R9) or (R10)CH3C(O)C(O)CH3 + hν351 nm → 2 CH3CO•
- Pulsed 248 nm photolysis of biacetyl (CH3C(O)C(O)CH3)/O2 mixtures:The same mechanism as above is utilized, but with a higher fraction of subsequent decomposition (R10)
- Pulsed 248 nm photolysis of acetone (CH3C(O)CH3)/O2 mixturesfollowed by (R5), (R9), and (R10).CH3C(O)CH3 + hν248 nm → CH3CO• + •CH3
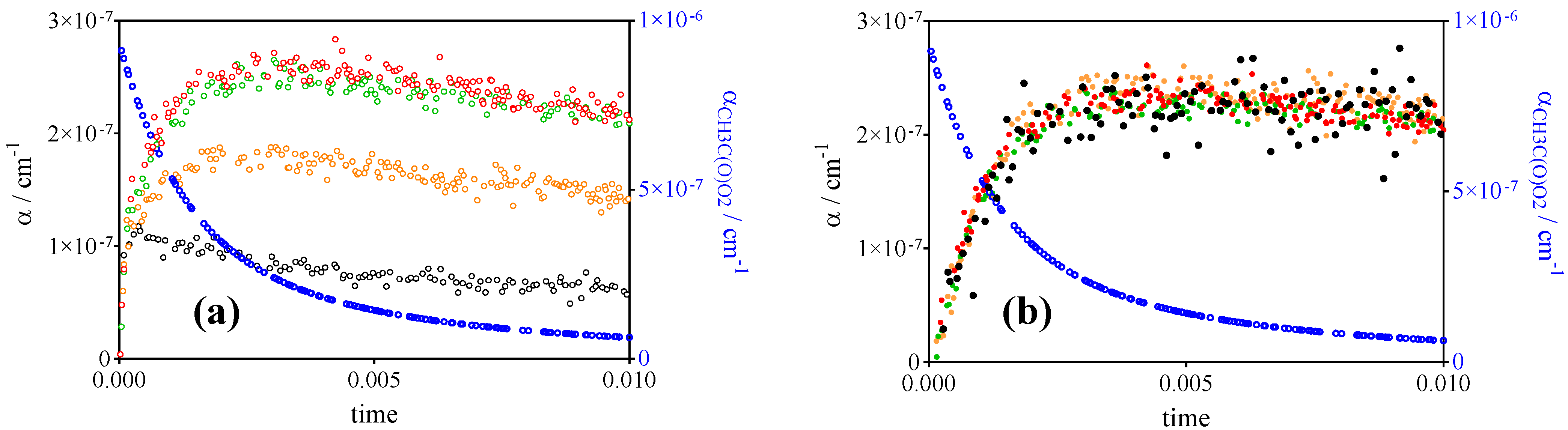
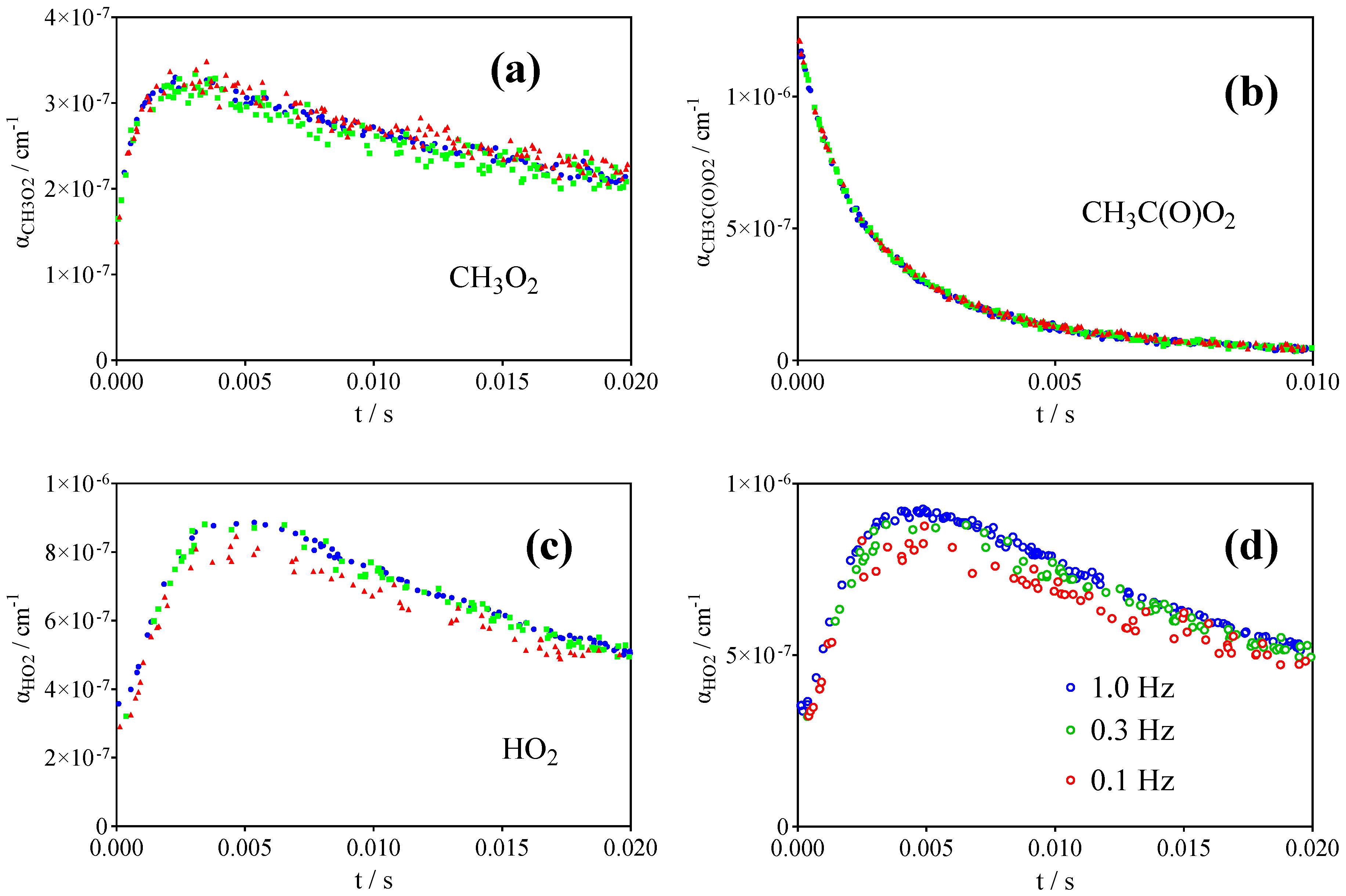
2.2. Quantification of CH3C(O)O2•
2.3. Quantification of HO2•
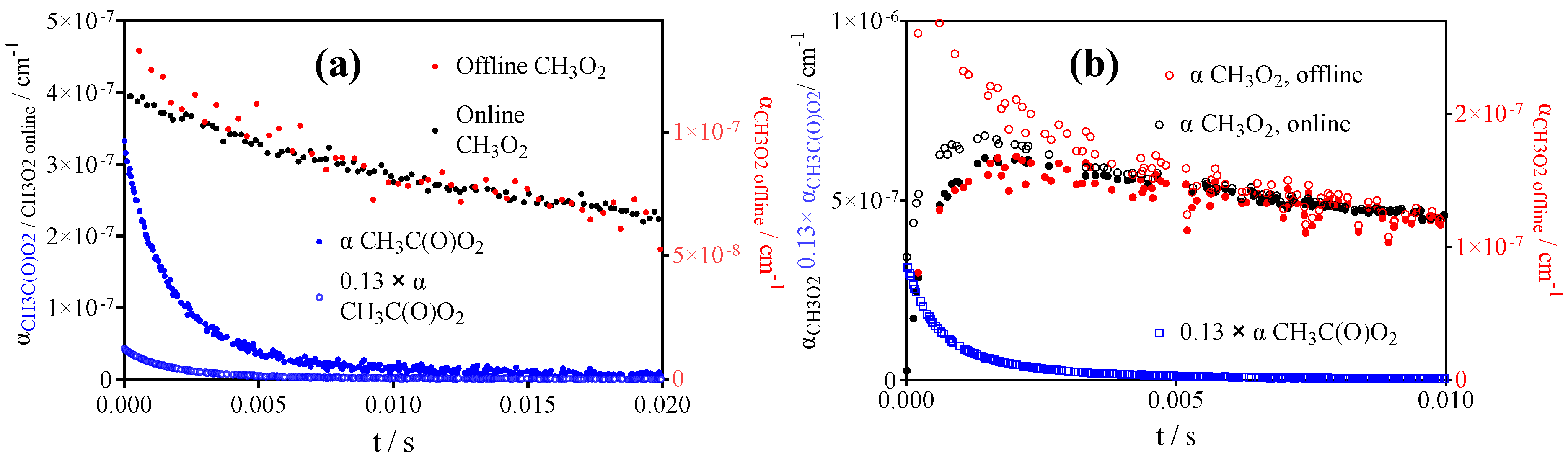
2.4. Quantification of CH3O2•
- -
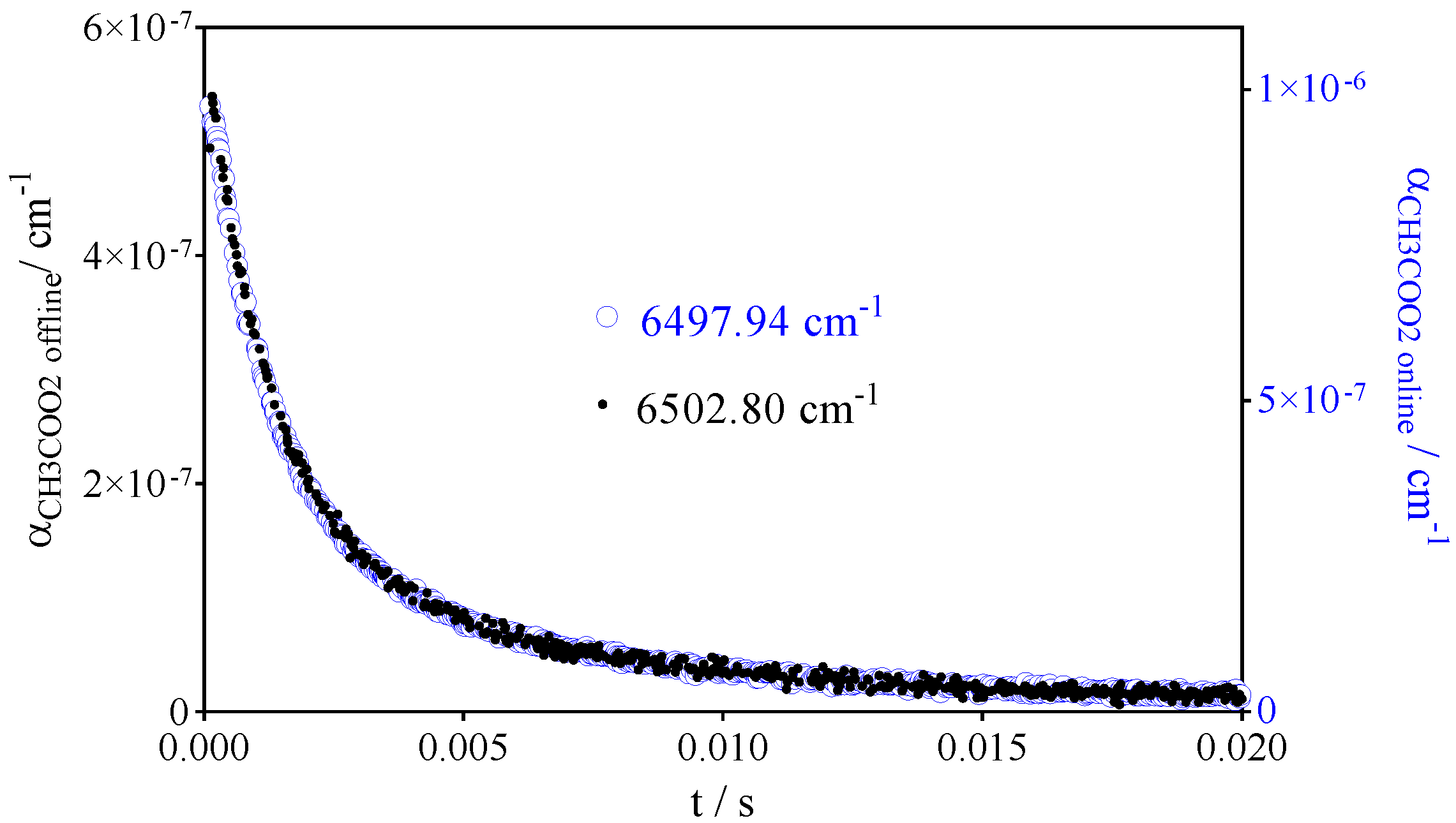
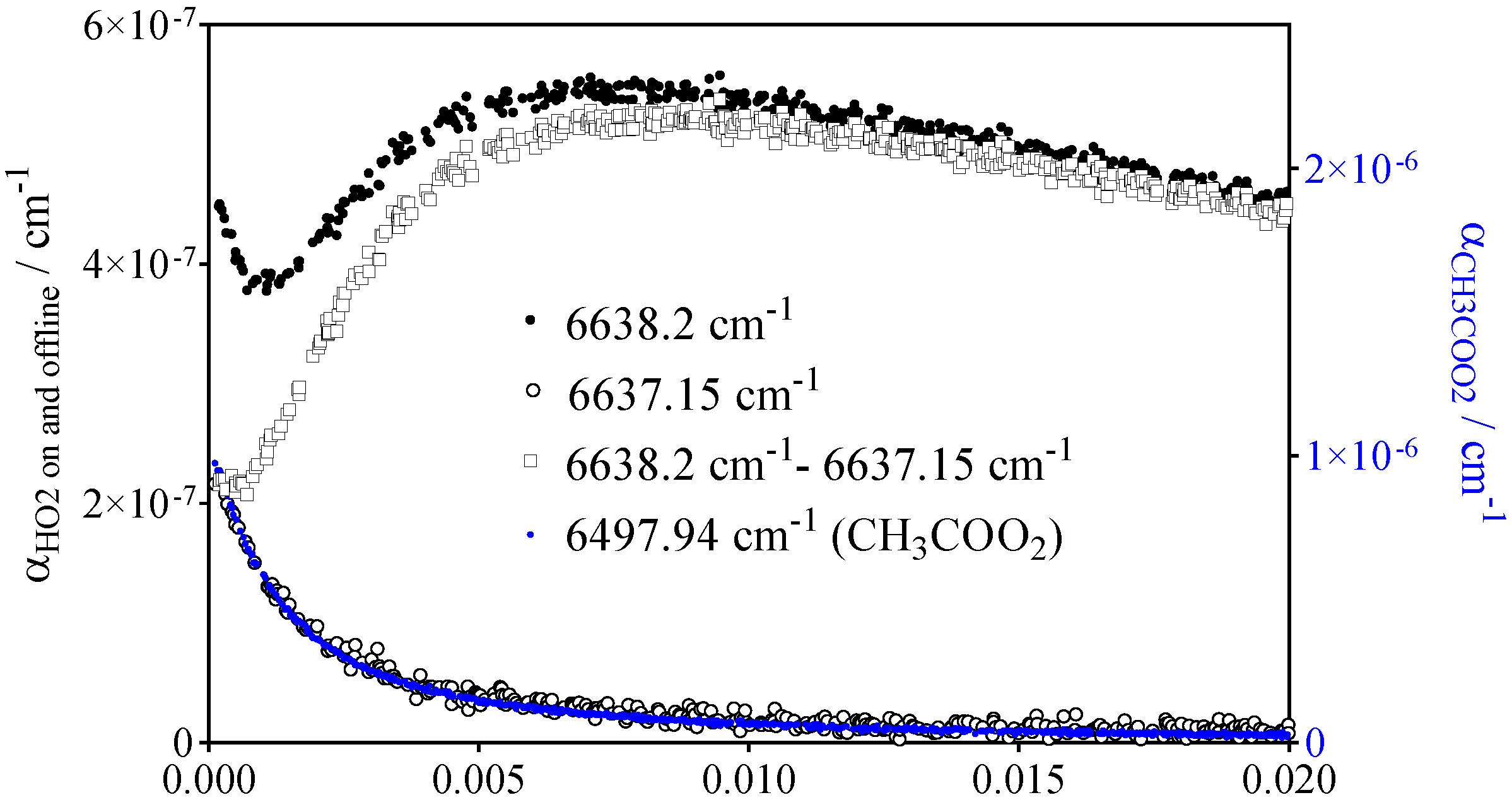
- CH3C(O)O2• at 6497.94 cm−1 with σ = 3.3 × 10−20 cm2
- -
- HO2• at 6638.2 cm−1 with σ = 2.0 × 10−19 cm2 at 100 Torr O2 and 2.72 × 10−19 cm2 at 50 Torr He after subtracting the offline signal measured at 6637.15 cm−1
- -
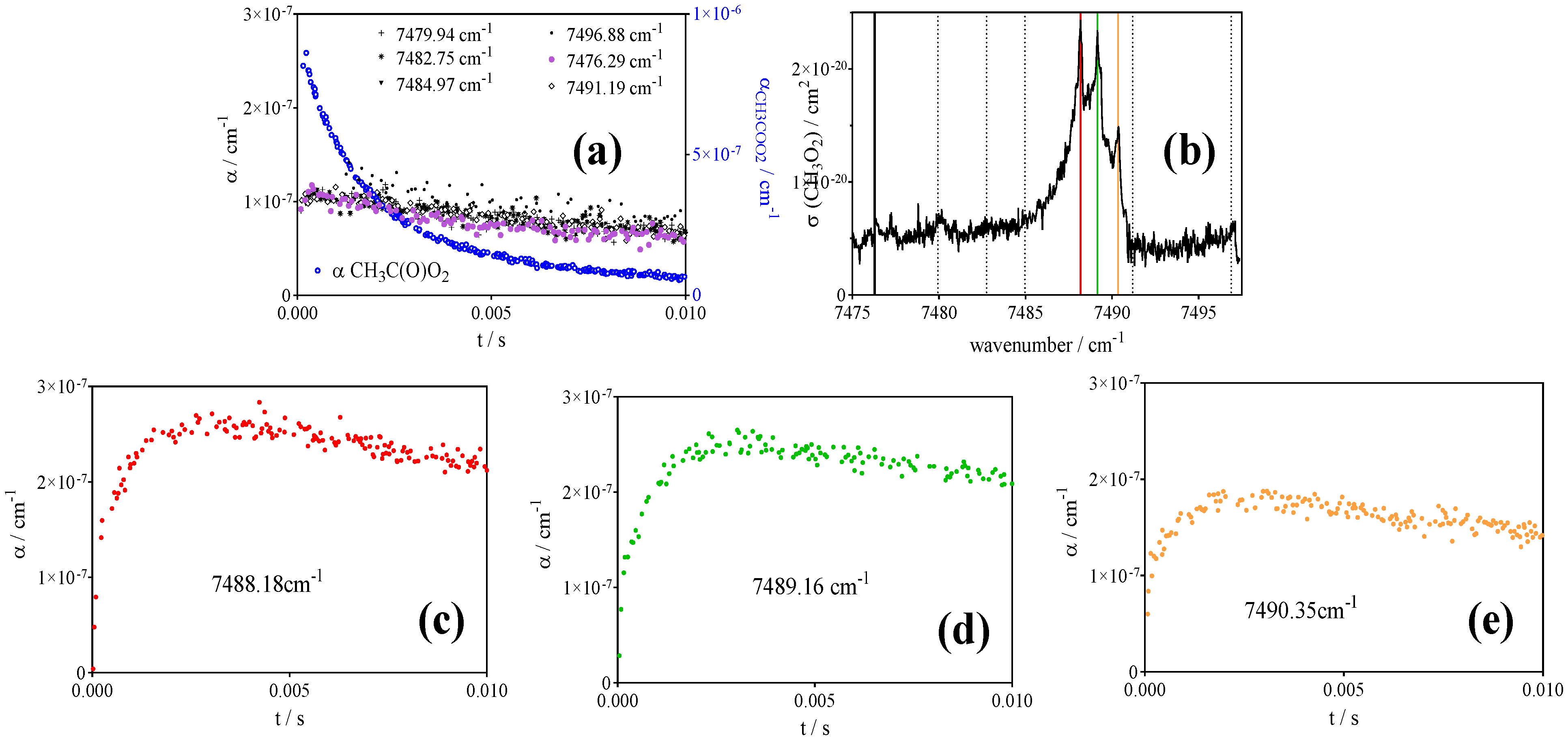
- CH3O2• at 7489.16 cm−1 with σ = 2.4 × 10−20 cm2 after subtracting 0.13 × αCH3C(O)O2 such as measured at 6497.94 cm−1 (or, for convenience, by representing [CH3O2•] as [CH3O2•] + 0.179 × [CH3C(O)O2•], with 0.179 = 4.3 × 10−21 cm2/2.4 × 10−20 cm2).
3. Results and Discussion
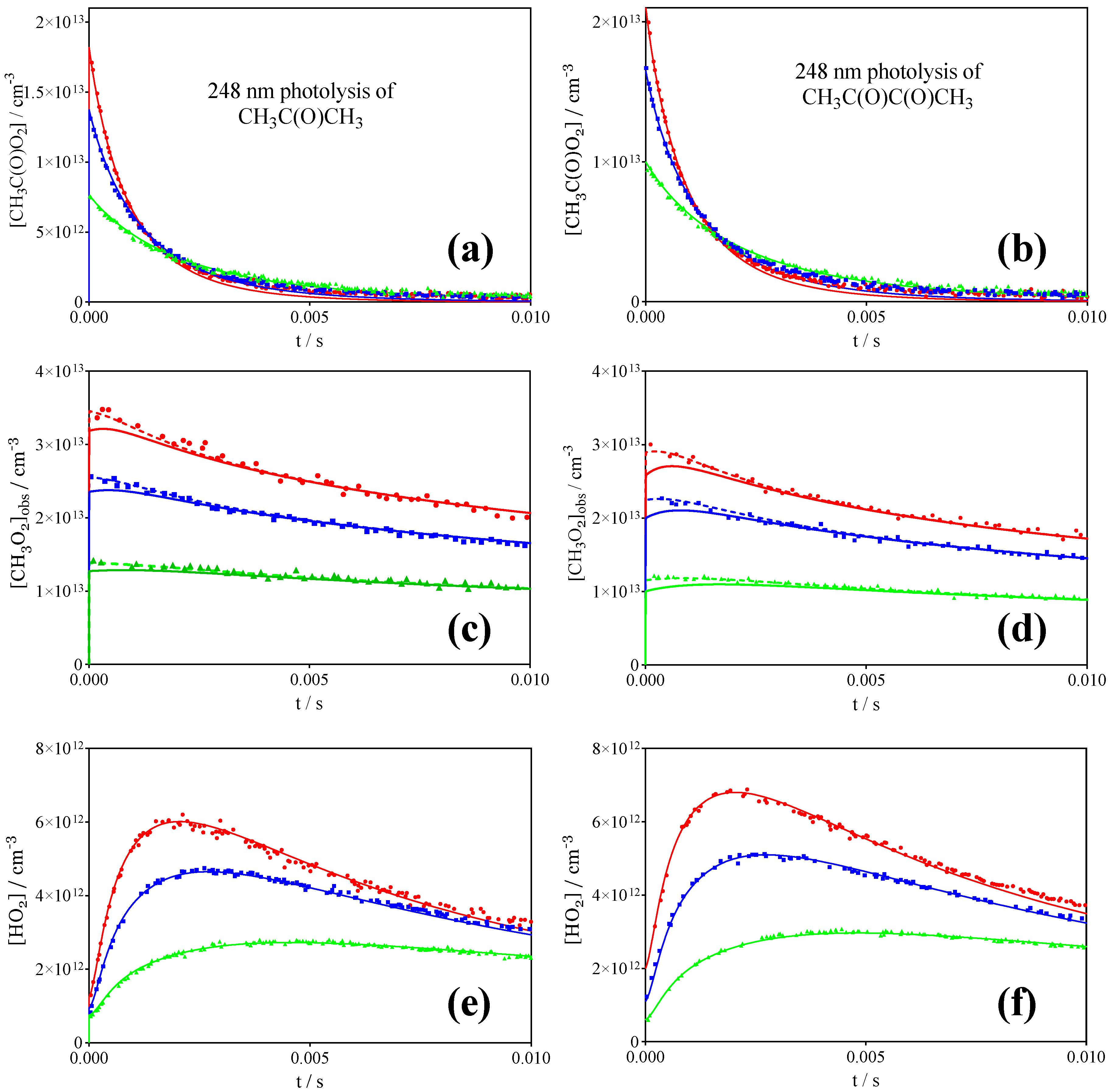
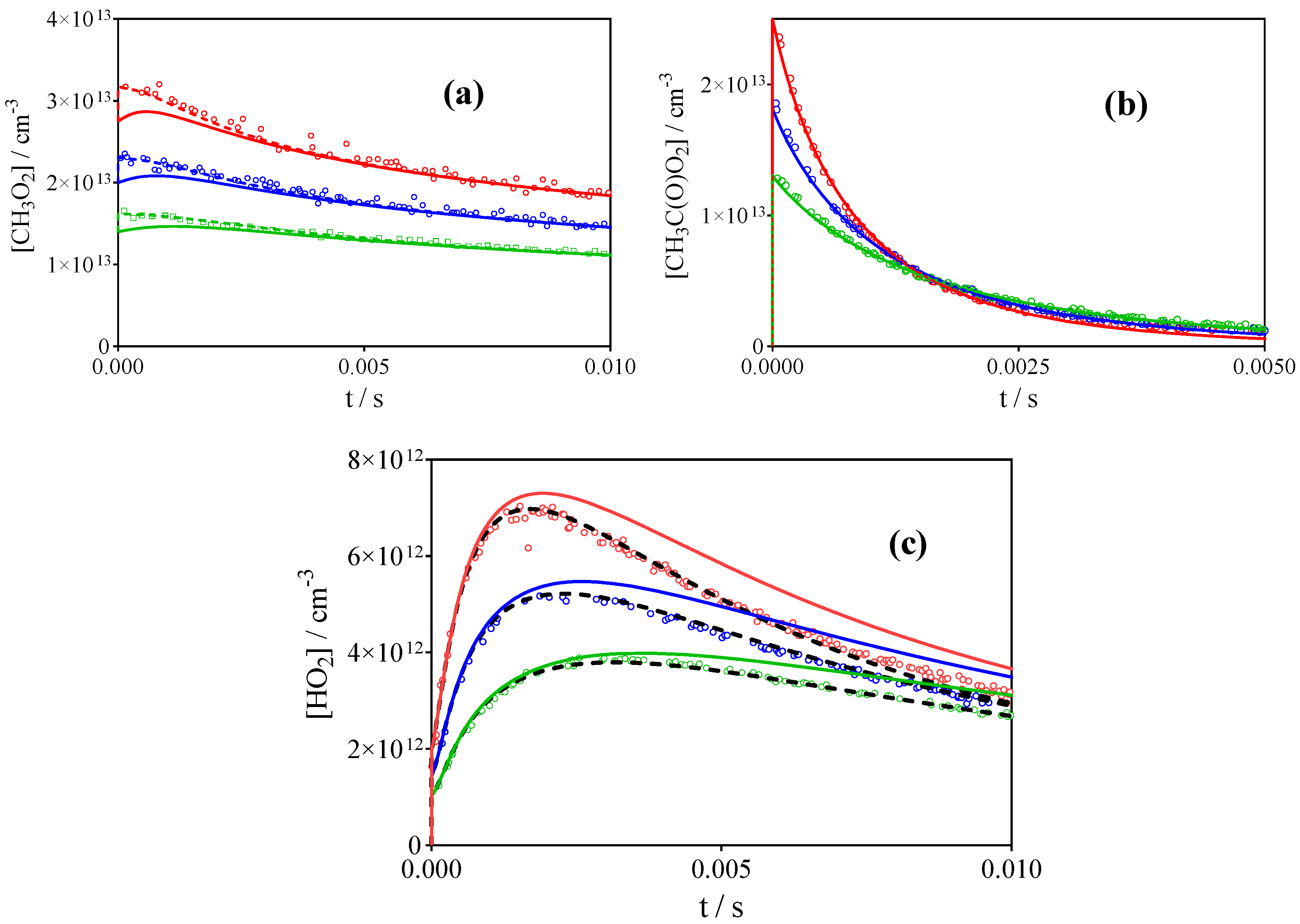
3.1. Photolysis of CH3C(O)CH3 and CH3C(O)C(O)CH3
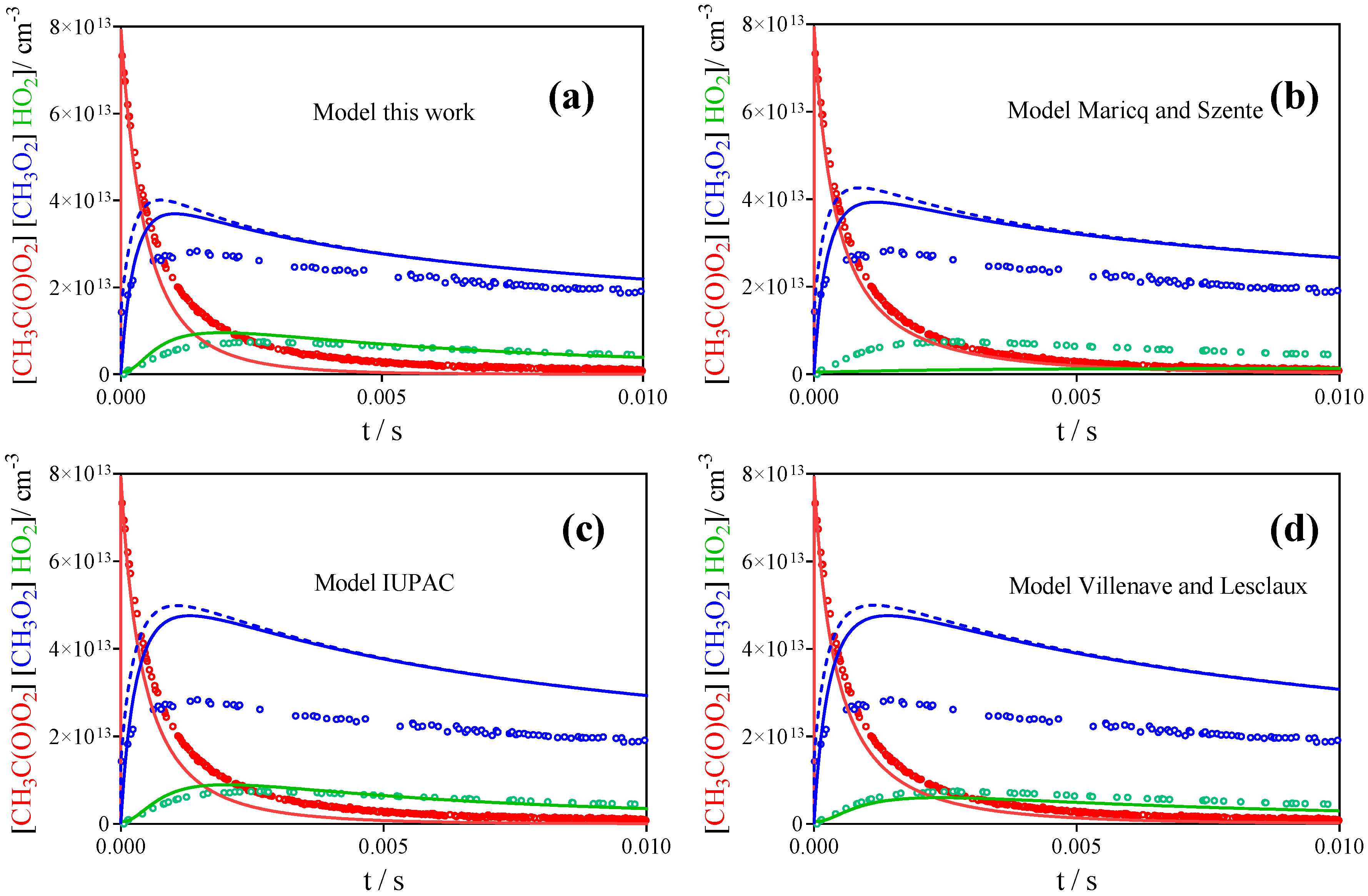
3.2. Comparison with Literature Rate Constants
3.3. Reaction of Cl-Atoms with CH3CHO as Precursor
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orlando, J.J.; Tyndall, G.S. Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev. 2012, 41, 6294–6317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fittschen, C. The reaction of peroxy radicals with OH radicals. Chem. Phys. Lett. 2019, 725, 102–108. [Google Scholar] [CrossRef]
- Assaf, E.; Song, B.; Tomas, A.; Schoemaecker, C.; Fittschen, C. Rate Constant of the Reaction between CH3O2 Radicals and OH Radicals revisited. J. Phys. Chem. A 2016, 120, 8923–8932. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.G.; Derwent, R.G.; Orlando, J.J.; Tyndall, G.S.; Wallington, T.J. Mechanisms of Atmospheric Oxidation of the Alkanes; Oxford University Press: New York, NY, USA, 2008. [Google Scholar]
- Guenther, A.B.; Jiang, X.; Heald, C.L.; Sakulyanontvittaya, T.; Duhl, T.; Emmons, L.K.; Wang, X. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2. 1): An extended and updated framework for modeling biogenic emissions. Geosci. Model Dev. 2012, 5, 1471–1492. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.V.; Jacob, D.J.; Yantosca, R.M.; Sulprizio, M.P.; Millet, D.B.; Mao, J.; Paulot, F.; Singh, H.B.; Roiger, A.; Ries, L.; et al. Atmospheric peroxyacetyl nitrate (PAN): A global budget and source attribution. Atmos. Chem. Phys. 2014, 14, 2679–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Faloona, I.; Simpas, J.B.; Brune, W.; Shepson, P.B.; Couch, T.L.; Sumner, A.L.; Carroll, M.A.; Thornberry, T.; Apel, E.; et al. HOx budgets in a deciduous forest: Results from the PROPHET summer 1998 campaign. J. Geophys. Res. Atmos. 2001, 106, 24407–24427. [Google Scholar] [CrossRef]
- Lelieveld, J.; Butler, T.M.; Crowley, J.N.; Dillon, T.J.; Fischer, H.; Ganzeveld, L.; Harder, H.; Lawrence, M.G.; Martinez, M.; Taraborrelli, D.; et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature 2008, 452, 737–740. [Google Scholar] [CrossRef]
- Hofzumahaus, A.; Rohrer, F.; Lu, K.; Bohn, B.; Brauers, T.; Chang, C.C.; Fuchs, H.; Holland, F.; Kita, K.; Kondo, Y.; et al. Amplified trace gas removal in the troposphere. Science 2009, 324, 1702–1704. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, G.M.; Thornton, J.A.; Bouvier-Brown, N.C.; Goldstein, A.H.; Park, J.H.; McKay, M.; Matross, D.M.; Mao, J.; Brune, W.H.; LaFranchi, B.W.; et al. The Chemistry of Atmosphere-Forest Exchange (CAFE) Model—Part 2: Application to BEARPEX-2007 observations. Atmos. Chem. Phys. 2011, 11, 1269–1294. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Ren, X.; Brune, W.H.; Van Duin, D.M.; Cohen, R.C.; Park, J.-H.; Goldstein, A.H.; Paulot, F.; Beaver, M.R.; Crounse, J.D.; et al. Insights into hydroxyl measurements and atmospheric oxidation in a California forest. Atmos. Chem. Phys. 2012, 12, 8009–8020. [Google Scholar] [CrossRef] [Green Version]
- Fittschen, C.; Al Ajami, M.; Batut, S.; Ferracci, V.; Archer-Nicholls, S.; Archibald, A.T.; Schoemaecker, C. ROOOH: A missing piece of the puzzle for OH measurements in low-NO environments? Atmos. Chem. Phys. 2019, 19, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Addison, M.C.; Burrows, J.P.; Cox, R.A.; Patrick, R. Absorption spectrum and kinetics of the acetylperoxy radical. Chem. Phys. Lett. 1980, 73, 283–287. [Google Scholar] [CrossRef]
- Moortgat, G.; Veyret, B.; Lesclaux, R. Absorption spectrum and kinetics of reactions of the acetylperoxy radicals. J. Phys. Chem. 1989, 93, 2362–2368. [Google Scholar] [CrossRef]
- Lightfoot, P.D.; Cox, R.A.; Crowley, J.N.; Destriau, M.; Hayman, G.D.; Jenkin, M.E.; Moortgat, G.K.; Zabel, F. Organic peroxy radicals—Kinetics, spectroscopy and tropospheric chemistry. Atmos. Environ. Part A-Gen. Top. 1992, 26, 1805–1961. [Google Scholar] [CrossRef]
- Roehl, C.M.; Bauer, D.; Moortgat, G.K. Absorption spectrum and kinetics of the acetylperoxy radical. J. Phys. Chem. 1996, 100, 4038–4047. [Google Scholar] [CrossRef]
- Maricq, M.M.; Szente, J.J. The CH3C(O)O2 Radical. Its UV Spectrum, Self-Reaction Kinetics, and Reaction with CH3O2. J. Phys. Chem 1996, 100, 4507–4513. [Google Scholar] [CrossRef]
- Niki, H.; Maker, P.D.; Savage, C.M.; Breitenbach, L.P. FTIR study of the kinetics and mechanism for chlorine-atom-initiated reactions of acetaldehyde. J. Phys. Chem. 1985, 89, 588–591. [Google Scholar] [CrossRef]
- Moortgat, G.K.; Veyret, B.; Lesclaux, R. Kinetics of the reaction of HO2 with CH3C(O)O2 in the temperature range 253–368 K. Chem. Phys. Lett. 1989, 160, 443–447. [Google Scholar] [CrossRef]
- Crawford, M.A.; Wallington, T.J.; Szente, J.J.; Maricq, M.M.; Francisco, J.S. Kinetics and Mechanism of the Acetylperoxy + HO2 Reaction. J. Phys. Chem. A 1999, 103, 365–378. [Google Scholar] [CrossRef]
- Tomas, A.; Villenave, E.; Lesclaux, R. Reactions of the HO2 Radical with CH3CHO and CH3C(O)O2 in the Gas Phase. J. Phys. Chem. A 2001, 105, 3505–3514. [Google Scholar] [CrossRef]
- LeCrane, J.-P.; Rayez, M.-T.; Rayez, J.-C.; Villenave, E. A reinvestigation of the kinetics and the mechanism of the CH3C(O)O2+ HO2 reaction using both experimental and theoretical approaches. Phys. Chem. Chem. Phys. 2006, 8, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Horie, O.; Moortgat, G.K. Reactions of CH3C(O)O2 radicals with CH3O2 and HO2 between 263 and 333 K. A product study. J. Chem. Soc. Faraday Trans. 1992, 88, 3305–3312. [Google Scholar] [CrossRef]
- Jenkin, M.E.; Hurley, M.D.; Wallington, T.J. Investigation of the radical product channel of the CH3C(O)O2 + HO2 reaction in the gas phase. Phys. Chem. Chem. Phys. 2007, 9, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Dillon, T.J.; Crowley, J.N. Direct Detection of OH Formation in the Reactions of HO2 with CH3C(O)O2 and Other Substituted Peroxy Radicals. Atmos. Chem. Phys. 2008, 8, 4877–4889. [Google Scholar] [CrossRef] [Green Version]
- Hasson, A.S.; Tyndall, G.S.; Orlando, J.J. A Product Yield Study of the Reaction of HO2 Radicals with Ethyl Peroxy (C2H5O2), Acetyl Peroxy (CH3C(O)O2), and Acetonyl Peroxy (CH3C(O)CH2O2) Radicals. J. Phys. Chem. A 2004, 108, 5979–5989. [Google Scholar] [CrossRef]
- Hui, A.O.; Fradet, M.; Okumura, M.; Sander, S.P. Temperature Dependence Study of the Kinetics and Product Yields of the HO2 + CH3C(O)O2 Reaction by Direct Detection of OH and HO2 Radicals Using 2f-IR Wavelength Modulation Spectroscopy. J. Phys. Chem. A 2019, 123, 3655–3671. [Google Scholar] [CrossRef] [Green Version]
- Villenave, E.; Lesclaux, R. Kinetics of the Cross Reactions of CH3O2 and C2H5O2 Radicals with Selected Peroxy Radicals. J. Phys. Chem. 1996, 100, 14372–14382. [Google Scholar] [CrossRef]
- Shallcross, D.E.; Raventos-Duran, M.T.; Bardwell, M.W.; Bacak, A.; Solman, Z.; Percival, C.J. A semi-empirical correlation for the rate coefficients for cross- and self-reactions of peroxy radicals in the gas-phase. Atmos. Environ. 2005, 39, 763–771. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume II—Gas Phase Reactions of Organic Species. Atmos. Chem. Phys. 2006, 6, 3625–4055. [Google Scholar] [CrossRef] [Green Version]
- Burkholder, J.B.; Sander, S.P.; Abbatt, J.P.; Barker, J.R.; Cappa, C.; Crounse, J.D.; Dibble, T.S.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19; JPL Publication: Pasadena, CA, USA, 2020. [Google Scholar]
- Shamas, M.; Assali, M.; Zhang, C.; Tang, X.; Zhang, W.; Pillier, L.; Schoemaecker, C.; Fittschen, C. Rate Constant and Branching Ratio for the Reactions of the Ethyl Peroxy Radical with Itself and with the Ethoxy Radical. ACS Earth Space Chem. 2022, 6, 181–188. [Google Scholar] [CrossRef]
- Thiebaud, J.; Aluculesei, A.; Fittschen, C. Formation of HO2 Radicals from the Photodissociation of H2O2 at 248 nm. J. Chem. Phys. 2007, 126, 186101. [Google Scholar] [CrossRef] [PubMed]
- Thiebaud, J.; Fittschen, C. Near Infrared cw-CRDS Coupled to Laser Photolysis: Spectroscopy and Kinetics of the HO2 Radical. Appl. Phys. B 2006, 85, 383–389. [Google Scholar] [CrossRef]
- Assaf, E.; Asvany, O.; Votava, O.; Batut, S.; Schoemaecker, C.; Fittschen, C. Measurement of line strengths in the à 2A’ ← X 2A” transition of HO2 and DO2. J. Quant. Spectrosc. Radiat. Transf. 2017, 201, 161–170. [Google Scholar] [CrossRef]
- Zhang, C.; Shamas, M.; Assali, M.; Tang, X.; Zhang, W.; Pillier, L.; Schoemaecker, C.; Fittschen, C. Absolute Absorption Cross-Section of the Ã←X~ Electronic Transition of the Ethyl Peroxy Radical and Rate Constant of Its Cross Reaction with HO2. Photonics 2021, 8, 296. [Google Scholar] [CrossRef]
- Parker, A.; Jain, C.; Schoemaecker, C.; Szriftgiser, P.; Votava, O.; Fittschen, C. Simultaneous, Time-Resolved Measurements of OH and HO2 Radicals by Coupling of High Repetition Rate LIF and cw-CRDS Techniques to a Laser Photolysis Reactor and its Application to the Photolysis of H2O2. Appl. Phys. B 2011, 103, 725–733. [Google Scholar] [CrossRef]
- Votava, O.; Masat, M.; Parker, A.E.; Jain, C.; Fittschen, C. Microcontroller Based Resonance Tracking unit for Time Resolved Continuous wave Cavity-Ringdown Spectroscopy Measurements. Rev. Sci. Instrum. 2012, 83, 043110. [Google Scholar] [CrossRef] [PubMed]
- Morajkar, P.; Bossolasco, A.; Schoemaecker, C.; Fittschen, C. Photolysis of CH3CHO at 248 nm: Evidence of Triple Fragmentation from Primary Quantum Yield of CH3 and HCO Radicals and H Atoms. J. Chem. Phys. 2014, 140, 214308. [Google Scholar] [CrossRef]
- Blitz, M.A.; Heard, D.E.; Pilling, M.J. OH formation from CH3CO+O2: A convenient experimental marker for the acetyl radical. Chem. Phys. Lett. 2002, 365, 374–379. [Google Scholar] [CrossRef]
- Zalyubovsky, S.J.; Glover, B.G.; Miller, T.A. Cavity Ringdown Spectroscopy of the à − Electronic Transition of the CH3C(O)O2 Radical. J. Phys. Chem. A 2003, 107, 7704–7712. [Google Scholar] [CrossRef]
- Rolletter, M.; Assaf, E.; Assali, M.; Fuchs, H.; Fittschen, C. The absorption spectrum and absolute absorption cross sections of acetylperoxy radicals, CH3C(O)O2 in the near IR. J. Quant. Spectrosc. Radiat. Transf. 2020, 245, 106877. [Google Scholar] [CrossRef]
- Thiebaud, J.; Crunaire, S.; Fittschen, C. Measurement of Line Strengths in the 2n1 Band of the HO2 Radical using Laser Photolysis / Continous wave Cavity Ring Down Spectroscopy (cw-CRDS). J. Phys. Chem. A 2007, 111, 6959–6966. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tyndall, G.S.; Orlando, J.J. Spectroscopic and Kinetic Properties of HO2 Radicals and the Enhancement of the HO2 Self Reaction by CH3OH and H2O. J. Phys. Chem. A 2010, 114, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.; Thiebaud, J.; Orphal, J.; Fittschen, C. Air-Broadening Coefficients of the HO2 Radical in the 2v1 Band Measured Using cw-CRDS. J. Mol. Spectrosc. 2007, 242, 64–69. [Google Scholar] [CrossRef]
- Assaf, E.; Liu, L.; Schoemaecker, C.; Fittschen, C. Absorption spectrum and absorption cross sections of the 2v1 band of HO2 between 20 and 760 Torr air in the range 6636 and 6639 cm−1. J. Quant. Spectrosc. Radiat. Transf. 2018, 211, 107–114. [Google Scholar] [CrossRef]
- Onel, L.; Brennan, A.; Gianella, M.; Ronnie, G.; Lawry Aguila, A.; Hancock, G.; Whalley, L.; Seakins, P.W.; Ritchie, G.A.D.; Heard, D.E. An intercomparison of HO2 measurements by fluorescence assay by gas expansion and cavity ring-down spectroscopy within HIRAC (Highly Instrumented Reactor for Atmospheric Chemistry). Atmos. Meas. Tech. 2017, 10, 4877–4894. [Google Scholar] [CrossRef] [Green Version]
- Assali, M.; Rakovsky, J.; Votava, O.; Fittschen, C. Experimental determination of the rate constants of the reactions of HO2 + DO2 and DO2 + DO2. Int. J. Chem. Kinet. 2020, 52, 197–206. [Google Scholar] [CrossRef]
- Thiebaud, J.; Thevenet, F.; Fittschen, C. OH Radicals and H2O2 Molecules in the Gas Phase near to TiO2 Surfaces. J. Phys. Chem. C 2010, 114, 3082–3088. [Google Scholar] [CrossRef]
- Faragó, E.P.; Viskolcz, B.; Schoemaecker, C.; Fittschen, C. Absorption Spectrum and Absolute Absorption Cross Sections of CH3O2 Radicals and CH3I Molecules in the Wavelength Range 7473–7497 cm−1. J. Phys. Chem. A 2013, 117, 12802–12811. [Google Scholar] [CrossRef]
- Atkinson, D.B.; Spillman, J.L. Alkyl Peroxy Radical Kinetics Measured Using Near-infrared CW-Cavity Ring-down Spectroscopy. J. Phys. Chem. A 2002, 106, 8891–8902. [Google Scholar] [CrossRef]
- Onel, L.; Brennan, A.; Gianella, M.; Hooper, J.; Ng, N.; Hancock, G.; Whalley, L.; Seakins, P.W.; Ritchie, G.A.D.; Heard, D.E. An intercomparison of CH3O2 measurements by Fluorescence Assay by Gas Expansion and Cavity Ring–Down Spectroscopy within HIRAC (Highly Instrumented Reactor for Atmospheric Chemistry). Atmos. Meas. Tech. 2017, 10, 4877–4894. [Google Scholar] [CrossRef] [Green Version]
- Jacquinet-Husson, N.; Armante, R.; Scott, N.A.; Chédin, A.; Crépeau, L.; Boutammine, C.; Bouhdaoui, A.; Crevoisier, C.; Capelle, V.; Boonne, C.; et al. The 2015 edition of the GEISA spectroscopic database. J. Mol. Spectrosc. 2016, 327, 31–72. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.H.; Ramsay, D.A. High-Resolution Study of the Ã2A’→X2A’’ Transition of HO2: Analysis of the 000–000 Band. J. Mol. Spectrosc. 1997, 185, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Miller, T. Private communication by e-mail about absorption spectrum of CH3C(O)O2. 2021. [Google Scholar]
- Delbos, E.; Fittschen, C.; Hippler, H.; Krasteva, N.; Olzmann, M.; Viskolcz, B. Rate Coefficients and Equilibrium Constant for the CH2CHO + O2 Reaction System. J. Phys. Chem. A 2006, 110, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Bartels, M.; Hoyermann, K.; Lange, U. An Experimental Study of the Reactions CH3CHO + Cl, C2H4O + Cl, and C2H4O + F in the Gas-Phase. Ber. Bunsenges. Phys. Chem. 1989, 93, 423–427. [Google Scholar] [CrossRef]
- Assaf, E.; Fittschen, C. Cross Section of OH Radical Overtone Transition near 7028 cm−1 and Measurement of the Rate Constant of the Reaction of OH with HO2 Radicals. J. Phys. Chem. A 2016, 120, 7051–7059. [Google Scholar] [CrossRef]
- Parker, A.; Jain, C.; Schoemaecker, C.; Fittschen, C. Kinetics of the Reaction of OH Radicals with CH3OH and CD3OD Studied by Laser Photolysis Coupled to High Repetition Rate Laser Induced Fluorescence. React. Kinet. Catal. Lett. 2009, 96, 291–297. [Google Scholar] [CrossRef]
- Fernandes, R.X.; Luther, K.; Troe, J. Falloff Curves for the Reaction CH3 + O2 (+ M) → CH3O2 (+ M) in the Pressure Range 2–1000 Bar and the Temperature Range 300–700 K. J. Phys. Chem. A 2006, 110, 4442–4449. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume 1—Gas Phase Reactions of Ox, HOx, NOx, and SOx, Species. Atmos. Chem. Phys. 2004, 4, 1461–1738. [Google Scholar] [CrossRef] [Green Version]
- Dagaut, P.; Wallington, T.J.; Liu, R.Z.; Kurylo, M.J. A kinetic investigation of the gas-phase reactions of hydroxyl radicals with cyclic ketones and diones: Mechanistic insights. J. Phys. Chem. 1988, 92, 4375–4377. [Google Scholar] [CrossRef]
- Assaf, E.; Tanaka, S.; Kajii, Y.; Schoemaecker, C.; Fittschen, C. Rate constants of the reaction of C2–C4 peroxy radicals with OH radicals. Chem. Phys. Lett. 2017, 684, 245–249. [Google Scholar] [CrossRef]
- Tyndall, G.S.; Orlando, J.J.; Kegley-Owen, C.S.; Wallington, T.J.; Hurley, M.D. Rate coefficients for the reactions of chlorine atoms with methanol and acetaldehyde. Int. J. Chem. Kinet. 1999, 31, 776–784. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | IUPAC [30] | Roehl et al. [16] | Maricq, Szente [17] | Villenave, Lesclaux [28] | This work | |
| Reaction | ||||||
| CH3C(O)O2• + CH3O2• | 1.1 × 10−11 | 9.8 × 10−12 | 1.0 × 10−11 | 8.6 × 10−12 | 2.0 × 10−11 | |
| (R2a): CH3O• + CH3C(O)O• + O2 | 9.9 × 10−12 | 8.8 × 10−12 | 0 | 5.3 × 10−12 | 1.3 × 10−11 | |
| (R2b): CH3C(O)OH + HCHO + O2 | 1.1 × 10−12 | 1.0 × 10−12 | 1 × 10−11 | 2.9 × 10−12 | 6.5 × 10−12 | |
| α (R2a)/(R2) | 0.9 | 0.9 | 0 | 0.65 | 0.67 | |
| References | IUPAC [30] | Roehl et al. [16] | Maricq, Szente [17] | Moortgat et al. [14] | This work | |
| Reaction | ||||||
| 2 CH3C(O)O2• → 2 CH3C(O)O• + O2 | 1.6 × 10−11 | 1.36 × 10−11 | 1.5 × 10−11 | 1.6 × 10−11 | 1.3 × 10−11 | |
| Reaction | % CH3C(O)O2• | % CH3O2• | k9b/k9 |
|---|---|---|---|
| CH3CHO + Cl• | 100 | 0 | 0.007 * |
| CH3C(O)C(O)CH3 + hν351 nm 100 Torr | 89 | 11 | 0.014 |
| CH3C(O)C(O)CH3 + hν248 nm 100 Torr | 47 | 53 | 0.079 |
| CH3C(O)C(O)CH3 + hν248 nm 200 Torr | 50 | 50 | 0.079 |
| CH3C(O)CH3 + hν248 nm 100 Torr | 38 | 62 | 0.064 |
| CH3C(O)CH3 + hν248 nm 200 Torr | 40 | 60 | 0.064 |
| No | Reaction | k/10−11 cm3s−1 | References |
|---|---|---|---|
| 1 | 2 CH3C(O)O2•→ → → 2 CH3O2• | 1.3 ± 0.3 * | This work |
| 2a | CH3C(O)O2• + CH3O2• → CH3O• + CH3C(O)O• + O2 | 1.35 ± 0.2 * | This work |
| 2b | → molecular products | 0.65 ± 0.2 * | This work |
| 3a/b | CH3C(O)O2• + HO2• → molecular products | 0.86 | [27] |
| 3c | → CH3C(O)O• + •OH + O2 | 0.86 | |
| 5 | •CH3 + O2 (+M) → CH3O2• (+M) | 0.035 | [60] |
| 6 | CH3O• + O2 → CH2O + HO2• | 1.9 × 10−4 | [30] |
| 9a | CH3CO• + O2 (+ M) → CH3C(O)O2• (+ M) | 0.7 | [30] |
| 9b | CH3CO• + O2 → •CH2CO + HO2•/CH2C(O)O• + •OH | Varied | see Table 2 |
| 10 | CH3CO• → •CH3 + CO | Varied | see Table 2 |
| 11 | 2 HO2• → H2O2 + O2 | 0.17 | [61] |
| 12a | 2 CH3O2• → 2 CH3O• + O2 | 0.013 | [30] |
| 12b | → CH3OH + CH2O + O2 | 0.022 | |
| 13 | CH3O2•+ HO2• → CH3OOH | 0.52 | [30] |
| 14 | •OH + CH3CHO → CH3CO• + H2O | 1.6 | [30] |
| 15 | •OH + CH3C(O)CH3 → CH2C(O)CH3 + H2O | 0.022 | [30] |
| 16 | •OH + CH3C(O)C(O)CH3 → CH2C(O)C(O)CH3 + H2O | 0.025 | [62] |
| 17 | •OH + CH3O2• → CH3O• + HO2• | 12 | [3] |
| 18 | •OH + CH3C(O)O2• → CH3C(O)O3H | 10 | [12,63] |
| 19 | Cl• + CH3CHO → CH3CO• + HCl | 7.2 | [30] |
| 20 | Cl2 + CH3CO• → CH3COCl + Cl | 4.3 | [64] |
| 21 | HO2•, CH3O2•, CH3C(O)O2• → diffusion | 1–2 s−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assali, M.; Fittschen, C. Rate Constants and Branching Ratios for the Self-Reaction of Acetyl Peroxy (CH3C(O)O2•) and Its Reaction with CH3O2. Atmosphere 2022, 13, 186. https://doi.org/10.3390/atmos13020186
Assali M, Fittschen C. Rate Constants and Branching Ratios for the Self-Reaction of Acetyl Peroxy (CH3C(O)O2•) and Its Reaction with CH3O2. Atmosphere. 2022; 13(2):186. https://doi.org/10.3390/atmos13020186
Chicago/Turabian StyleAssali, Mohamed, and Christa Fittschen. 2022. "Rate Constants and Branching Ratios for the Self-Reaction of Acetyl Peroxy (CH3C(O)O2•) and Its Reaction with CH3O2" Atmosphere 13, no. 2: 186. https://doi.org/10.3390/atmos13020186
APA StyleAssali, M., & Fittschen, C. (2022). Rate Constants and Branching Ratios for the Self-Reaction of Acetyl Peroxy (CH3C(O)O2•) and Its Reaction with CH3O2. Atmosphere, 13(2), 186. https://doi.org/10.3390/atmos13020186