
Air Pollution and the Airways: Lessons from a Century of Human Urbanization

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Definition of Air Pollution
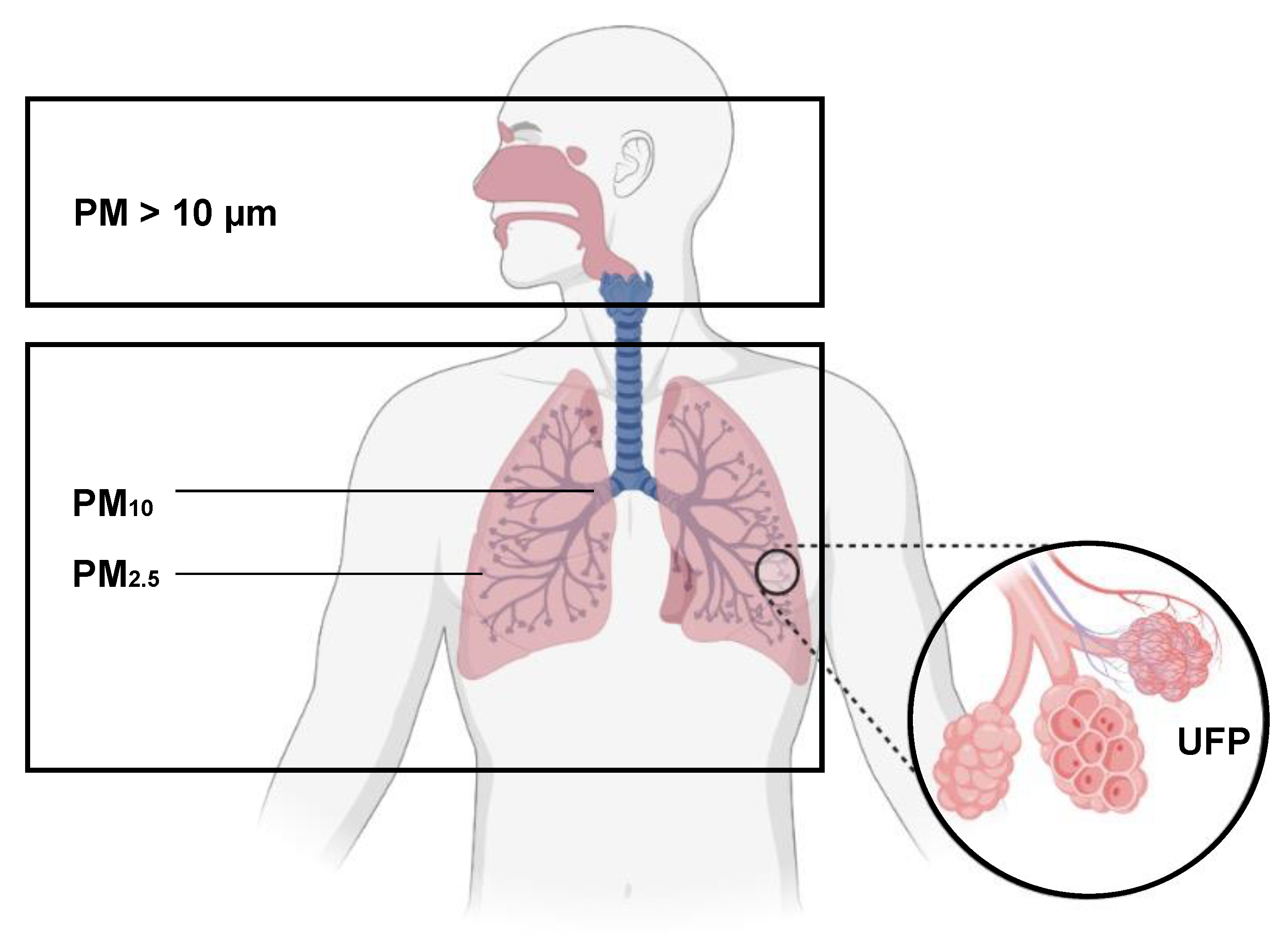
3. The Respiratory System and Air Pollution
4. Lessons Learned from Exposure Models: Murine and Human Data
4.1. Ozone
4.2. Carbon Monoxide
4.3. Carbon Dioxide
4.4. Volatile Organic Compounds and Polycyclic Aromatic Hydrocarbons
4.5. Particulate Matter
4.6. Diesel Exhaust Particles
4.7. Take Home Message from In Vivo Disease Models, Human and In Vitro Studies
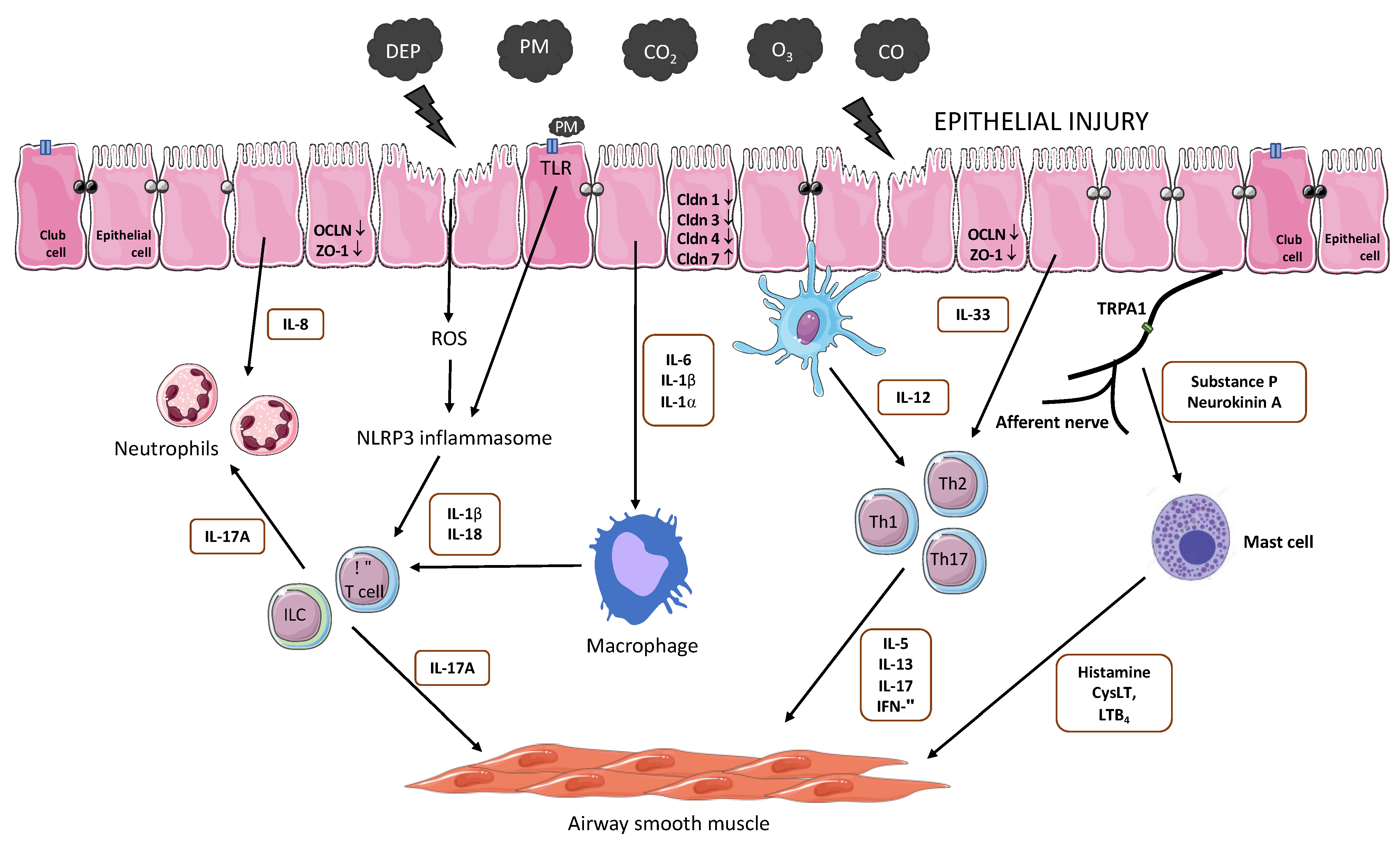
5. Pollution and Airway Diseases: Cause or Consequence?
5.1. Air Pollution and Asthma
5.2. Air Pollution and COPD
5.3. Air Pollution, Exercise and Bronchial Obstructive Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GHO | by Category | Deaths—By Country. Available online: https://apps.who.int/gho/data/node.main.BODAMBIENTAIRDTHS?lang=en (accessed on 6 April 2021).
- Ambient (Outdoor) Air Pollution. Available online: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health (accessed on 6 April 2021).
- Lelieveld, J.; Pozzer, A.; Pöschl, U.; Fnais, M.; Haines, A.; Münzel, T. Loss of Life Expectancy from Air Pollution Compared to Other Risk Factors: A Worldwide Perspective. Cardiovasc. Res. 2020, 116, 1910–1917. [Google Scholar] [CrossRef]
- Air Pollution. Available online: https://www.who.int/news-room/air-pollution (accessed on 29 April 2021).
- Tiotiu, A.I.; Novakova, P.; Nedeva, D.; Chong-Neto, H.J.; Novakova, S.; Steiropoulos, P.; Kowal, K. Impact of Air Pollution on Asthma Outcomes. Int. J. Environ. Res. Public Health 2020, 17, 6212. [Google Scholar] [CrossRef] [PubMed]
- SDG Indicator 11.6.2 Concentrations of Fine Particulate Matter (PM2.5). Available online: https://www.who.int/data/gho/data/indicators/indicator-details/GHO/concentrations-of-fine-particulate-matter-(pm2-5) (accessed on 6 April 2021).
- Zawar-Reza, P.; Spronken-Smith, R. Air Pollution Climatology. In Encyclopedia of World Climatology; Oliver, J.E., Ed.; Springer: Dordrecht, The Netherlands, 2005; pp. 21–32. ISBN 978-1-4020-3266-0. [Google Scholar]
- WHO Regional Publications; World Health Organization, Regional Office for Europe (Eds.) Glossary on Air Pollution; Obtainable from WHO Publications Centre: Copenhagen, Denmark; Albany, NY, USA, 1980; ISBN 978-92-9020-109-0. [Google Scholar]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieder, R.; Benbi, D.K. Reactive Nitrogen Compounds and Their Influence on Human Health: An Overview. Rev. Environ. Health 2021. [Google Scholar] [CrossRef]
- Bhatia, V.; Elnagary, L.; Dakshinamurti, S. Tracing the Path of Inhaled Nitric Oxide: Biological Consequences of Protein Nitrosylation. Pediatric Pulmonol. 2021, 56, 525–538. [Google Scholar] [CrossRef]
- Rouadi, P.W.; Idriss, S.A.; Naclerio, R.M.; Peden, D.B.; Ansotegui, I.J.; Canonica, G.W.; Gonzalez-Diaz, S.N.; Rosario Filho, N.A.; Ivancevich, J.C.; Hellings, P.W.; et al. Immunopathological Features of Air Pollution and Its Impact on Inflammatory Airway Diseases (IAD). World Allergy Organ. J. 2020, 13, 100467. [Google Scholar] [CrossRef] [PubMed]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3rd ed.; Wiley: Hoboken, NJ, USA, 2016; ISBN 978-1-118-94740-1. [Google Scholar]
- Meyerholz, D.K.; Suarez, C.J.; Dintzis, S.M.; Frevert, C.W. 9—Respiratory System. In Comparative Anatomy and Histology, 2nd ed.; Treuting, P.M., Dintzis, S.M., Montine, K.S., Eds.; Academic Press: San Diego, CA, USA, 2018; pp. 147–162. ISBN 978-0-12-802900-8. [Google Scholar]
- Gibson, P.G.; Simpson, J.L. The Overlap Syndrome of Asthma and COPD: What Are Its Features and How Important Is It? Thorax 2009, 64, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Labaki, W.W.; Han, M.K. Chronic Respiratory Diseases: A Global View. Lancet Respir. Med. 2020, 8, 531–533. [Google Scholar] [CrossRef]
- Kodros, J.K.; Volckens, J.; Jathar, S.H.; Pierce, J.R. Ambient Particulate Matter Size Distributions Drive Regional and Global Variability in Particle Deposition in the Respiratory Tract. GeoHealth 2018, 2, 298–312. [Google Scholar] [CrossRef] [Green Version]
- Glencross, D.A.; Ho, T.-R.; Camiña, N.; Hawrylowicz, C.M.; Pfeffer, P.E. Air Pollution and Its Effects on the Immune System. Free Radic. Biol. Med. 2020, 151, 56–68. [Google Scholar] [CrossRef]
- Losacco, C.; Perillo, A. Particulate Matter Air Pollution and Respiratory Impact on Humans and Animals. Environ. Sci. Pollut Res. 2018, 25, 33901–33910. [Google Scholar] [CrossRef]
- Lippmann, M.; Yeates, D.B.; Albert, R.E. Deposition, Retention, and Clearance of Inhaled Particles. Br. J. Ind. Med. 1980, 37, 337–362. [Google Scholar] [CrossRef] [Green Version]
- Geiser, M.; Kreyling, W.G. Deposition and Biokinetics of Inhaled Nanoparticles. Part Fibre Toxicol. 2010, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.M.; Loxham, M. Particulate Matter and the Airway Epithelium: The Special Case of the Underground? Eur. Respir. Rev. 2019, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaudel, C.; Fauconnier, L.; Julé, Y.; Ryffel, B. Functional and Morphological Differences of the Lung upon Acute and Chronic Ozone Exposure in Mice. Sci. Rep. 2018, 8, 10611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowska, M.; Quesniaux, V.F.J.; Akdis, C.A.; Chung, K.F.; Ryffel, B.; Togbe, D. Acute Respiratory Barrier Disruption by Ozone Exposure in Mice. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, J.; Zhang, H.; Shi, C.; Li, G.; Peng, Z.; Ma, J.; Zhou, Y.; Zhang, L. Short-Term Exposure to Ambient Air Pollution and Asthma Mortality. Am. J. Respir Crit. Care Med. 2019, 200, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.-G.; Lee, P.-H.; Lee, S.-H.; Park, C.-S.; Jang, A.-S. Impact of Ozone on Claudins and Tight Junctions in the Lungs. Environ. Toxicol. 2018, 33, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.A.; Laskin, D.L.; Smith, C.V.; Robertson, F.M.; Allen, E.M.G.; Doorn, J.A.; Slikker, W. Nitrative and Oxidative Stress in Toxicology and Disease. Toxicol. Sci. 2009, 112, 4–16. [Google Scholar] [CrossRef]
- Michaudel, C.; Couturier-Maillard, A.; Chenuet, P.; Maillet, I.; Mura, C.; Couillin, I.; Gombault, A.; Quesniaux, V.F.; Huaux, F.; Ryffel, B. Inflammasome, IL-1 and Inflammation in Ozone-Induced Lung Injury. Am. J. Clin. Exp. Immunol. 2016, 5, 33–40. [Google Scholar]
- Michaudel, C.; Maillet, I.; Fauconnier, L.; Quesniaux, V.; Chung, K.F.; Wiegman, C.; Peter, D.; Ryffel, B. Interleukin-1α Mediates Ozone-Induced Myeloid Differentiation Factor-88-Dependent Epithelial Tissue Injury and Inflammation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Wang, L.; Wang, M.; Wang, H.; Zhang, H.; Chen, Y.; Wang, X.; Gong, J.; Zhang, J.; Adcock, I.M.; et al. Mitochondrial ROS and NLRP3 Inflammasome in Acute Ozone-Induced Murine Model of Airway Inflammation and Bronchial Hyperresponsiveness. Free Radic. Res. 2019, 53, 780–790. [Google Scholar] [CrossRef]
- Che, L.; Jin, Y.; Zhang, C.; Lai, T.; Zhou, H.; Xia, L.; Tian, B.; Zhao, Y.; Liu, J.; Wu, Y.; et al. Ozone-Induced IL-17A and Neutrophilic Airway Inflammation Is Orchestrated by the Caspase-1-IL-1 Cascade. Sci. Rep. 2016, 6, 18680. [Google Scholar] [CrossRef] [Green Version]
- Enweasor, C.; Flayer, C.H.; Haczku, A. Ozone-Induced Oxidative Stress, Neutrophilic Airway Inflammation, and Glucocorticoid Resistance in Asthma. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, M.; Wang, M.; Wang, L.; Wang, H.; Zhang, H.; Chen, Y.; Gong, J.; Zhang, J.; Adcock, I.M.; et al. Roles of Mitochondrial ROS and NLRP3 Inflammasome in Multiple Ozone-Induced Lung Inflammation and Emphysema. Respir. Res. 2018, 19, 230. [Google Scholar] [CrossRef] [PubMed]
- Michaudel, C.; Bataille, F.; Maillet, I.; Fauconnier, L.; Colas, C.; Sokol, H.; Straube, M.; Couturier-Maillard, A.; Dumoutier, L.; van Snick, J.; et al. Ozone-Induced Aryl Hydrocarbon Receptor Activation Controls Lung Inflammation via Interleukin-22 Modulation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Dixon, A.E. The Effect of Obesity on Lung Function. Expert Rev. Respir. Med. 2018, 12, 755–767. [Google Scholar] [CrossRef]
- Clougherty, J.E. A Growing Role for Gender Analysis in Air Pollution Epidemiology. Environ. Health Perspect 2010, 118, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, S.A. Mechanistic Basis for Obesity-Related Increases in Ozone-Induced Airway Hyperresponsiveness in Mice. Ann. Ats 2017, 14, S357–S362. [Google Scholar] [CrossRef]
- Mathews, J.A.; Krishnamoorthy, N.; Kasahara, D.I.; Hutchinson, J.; Cho, Y.; Brand, J.D.; Williams, A.S.; Wurmbrand, A.P.; Ribeiro, L.; Cuttitta, F.; et al. Augmented Responses to Ozone in Obese Mice Require IL-17A and Gastrin-Releasing Peptide. Am. J. Respir. Cell Mol. Biol. 2017, 58, 341–351. [Google Scholar] [CrossRef]
- Osgood, R.S.; Kasahara, D.I.; Tashiro, H.; Cho, Y.; Shore, S.A. Androgens Augment Pulmonary Responses to Ozone in Mice. Physiol. Rep. 2019, 7, e14214. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Nicoleau, M.; Cabello, N.; Montes, D.; Zomorodi, N.; Chroneos, Z.C.; Silveyra, P. 17β-Estradiol Affects Lung Function and Inflammation Following Ozone Exposure in a Sex-Specific Manner. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L702–L716. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Cabello, N.; Nicoleau, M.; Chroneos, Z.C.; Silveyra, P. Modulation of the Lung Inflammatory Response to Ozone by the Estrous Cycle. Physiol. Rep. 2019, 7, e14026. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.; Cyphert-Daly, J.; Cumming, R.I.; Yu, Y.-R.; Gowdy, K.M.; Que, L.G.; Tighe, R.M. Sex Modifies Acute Ozone-Mediated Airway Physiologic Responses. Toxicol. Sci. 2019, 169, 499–510. [Google Scholar] [CrossRef]
- Cho, Y.; Abu-Ali, G.; Tashiro, H.; Brown, T.A.; Osgood, R.S.; Kasahara, D.I.; Huttenhower, C.; Shore, S.A. Sex Differences in Pulmonary Responses to Ozone in Mice. Role of the Microbiome. Am. J. Respir. Cell Mol. Biol. 2019, 60, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Last, J.A.; Ward, R.; Temple, L.; Kenyon, N.J. Ovalbumin-Induced Airway Inflammation and Fibrosis in Mice Also Exposed to Ozone. Inhal. Toxicol. 2004, 16, 33–43. [Google Scholar] [CrossRef]
- Liang, L.; Li, F.; Bao, A.; Zhang, M.; Chung, K.F.; Zhou, X. Activation of P38 Mitogen-Activated Protein Kinase in Ovalbumin and Ozone-Induced Mouse Model of Asthma. Respirology 2013, 18 (Suppl. 3), 20–29. [Google Scholar] [CrossRef]
- Larsen, S.T.; Matsubara, S.; McConville, G.; Poulsen, S.S.; Gelfand, E.W. Ozone Increases Airway Hyperreactivity and Mucus Hyperproduction in Mice Previously Exposed to Allergen. J. Toxicol. Environ. Health A 2010, 73, 738–747. [Google Scholar] [CrossRef]
- Bao, W.; Zhang, Y.; Zhang, M.; Bao, A.; Fei, X.; Zhang, X.; Zhou, X. Effects of Ozone Repeated Short Exposures on the Airway/Lung Inflammation, Airway Hyperresponsiveness and Mucus Production in a Mouse Model of Ovalbumin-Induced Asthma. Biomed. Pharmacother. 2018, 101, 293–303. [Google Scholar] [CrossRef]
- Hansen, J.S.; Nørgaard, A.W.; Koponen, I.K.; Sørli, J.B.; Paidi, M.D.; Hansen, S.W.K.; Clausen, P.A.; Nielsen, G.D.; Wolkoff, P.; Larsen, S.T. Limonene and Its Ozone-Initiated Reaction Products Attenuate Allergic Lung Inflammation in Mice. J. Immunotoxicol. 2016, 13, 793–803. [Google Scholar] [CrossRef]
- Bromberg, P.A. Mechanisms of the Acute Effects of Inhaled Ozone in Humans. Biochim. Biophys. Acta 2016, 1860, 2771–2781. [Google Scholar] [CrossRef]
- Cheng, W.; Duncan, K.E.; Ghio, A.J.; Ward-Caviness, C.; Karoly, E.D.; Diaz-Sanchez, D.; Conolly, R.B.; Devlin, R.B. Changes in Metabolites Present in Lung-Lining Fluid Following Exposure of Humans to Ozone. Toxicol. Sci. 2018, 163, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Arjomandi, M.; Balmes, J.R.; Frampton, M.W.; Bromberg, P.; Rich, D.Q.; Stark, P.; Alexis, N.E.; Costantini, M.; Hollenbeck-Pringle, D.; Dagincourt, N.; et al. Respiratory Responses to Ozone Exposure. MOSES (The Multicenter Ozone Study in Older Subjects). Am. J. Respir. Crit. Care Med. 2018, 197, 1319–1327. [Google Scholar] [CrossRef]
- Mirowsky, J.E.; Dailey, L.A.; Devlin, R.B. Differential Expression of Pro-Inflammatory and Oxidative Stress Mediators Induced by Nitrogen Dioxide and Ozone in Primary Human Bronchial Epithelial Cells. Inhal. Toxicol. 2016, 28, 374–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.-Z.; Zhou, Y.-B.; Zhou, L.-F.; Fu, Z.-D.; Wu, Y.-S.; Chen, Y.; Li, S.-N.; Huang, J.-R.; Li, J.-H. TRPC6 Modulates Adhesion of Neutrophils to Airway Epithelial Cells via NF-ΚB Activation and ICAM-1 Expression with Ozone Exposure. Exp. Cell Res. 2019, 377, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.D.; Ivins, S.; Alexis, N.E.; Wu, J.; Bromberg, P.A.; Brar, S.S.; Travlos, G.; London, S.J. Effect of Obesity on Acute Ozone-Induced Changes in Airway Function, Reactivity, and Inflammation in Adult Females. PLoS ONE 2016, 11, e0160030. [Google Scholar] [CrossRef]
- Bleecker, M.L. Chapter 12—Carbon monoxide intoxication. In Handbook of Clinical Neurology; Lotti, M., Bleecker, M.L., Eds.; Occupational Neurology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 131, pp. 191–203. [Google Scholar]
- Hampson, N.B. Carboxyhemoglobin: A Primer for Clinicians. Undersea Hyperb. Med. 2018, 45, 165–171. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic Obstructive Pulmonary Disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Craig, J.M.; Scott, A.L.; Mitzner, W. Immune-Mediated Inflammation in the Pathogenesis of Emphysema: Insights from Mouse Models. Cell Tissue Res. 2017, 367, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Laucho-Contreras, M.E.; Taylor, K.L.; Mahadeva, R.; Boukedes, S.S.; Owen, C.A. Automated Measurement of Pulmonary Emphysema and Small Airway Remodeling in Cigarette Smoke-Exposed Mice. JoVE 2015, e52236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serré, J.; Tanjeko, A.T.; Mathyssen, C.; Vanherwegen, A.-S.; Heigl, T.; Janssen, R.; Verbeken, E.; Maes, K.; Vanaudenaerde, B.; Janssens, W.; et al. Enhanced Lung Inflammatory Response in Whole-Body Compared to Nose-Only Cigarette Smoke-Exposed Mice. Respir. Res. 2021, 22, 86. [Google Scholar] [CrossRef]
- Kogel, U.; Wong, E.T.; Szostak, J.; Tan, W.T.; Lucci, F.; Leroy, P.; Titz, B.; Xiang, Y.; Low, T.; Wong, S.K.; et al. Impact of Whole-Body versus Nose-Only Inhalation Exposure Systems on Systemic, Respiratory, and Cardiovascular Endpoints in a 2-Month Cigarette Smoke Exposure Study in the ApoE−/− Mouse Model. J. Appl. Toxicol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Li, D.; Ouyang, H.; Huang, J.; Long, Z.; Liang, Z.; Chen, Y.; Chen, Y.; Zheng, Q.; Kuang, M.; et al. Comparison and Evaluation of Two Different Methods to Establish the Cigarette Smoke Exposure Mouse Model of COPD. Sci. Rep. 2017, 7, 15454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauderly, J.L.; Bechtold, W.E.; Bond, J.A.; Brooks, A.L.; Chen, B.T.; Cuddihy, R.G.; Harkema, J.R.; Henderson, R.F.; Johnson, N.F.; Rithidech, K.; et al. Comparison of 3 Methods of Exposing Rats to Cigarette Smoke. Exp. Pathol. 1989, 37, 194–197. [Google Scholar] [CrossRef]
- Milad, N.; Pineault, M.; Lechasseur, A.; Routhier, J.; Beaulieu, M.-J.; Aubin, S.; Morissette, M.C. Neutrophils and IL-1α Regulate Surfactant Homeostasis during Cigarette Smoking. J. Immunol. 2021, 206, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, C.D.; Yang, Y.R.; Qin, X.F.; Wang, J.J.; Zhang, X.; Du, X.N.; Yang, X.; Wang, Y.; Li, L.; et al. Role of the IL-33/ST2 Axis in Cigarette Smoke-Induced Airways Remodelling in Chronic Obstructive Pulmonary Disease. Thorax 2021. [Google Scholar] [CrossRef]
- Guerrina, N.; Traboulsi, H.; Rico de Souza, A.; Bossé, Y.; Thatcher, T.H.; Robichaud, A.; Ding, J.; Li, P.Z.; Simon, L.; Pareek, S.; et al. Aryl Hydrocarbon Receptor Deficiency Causes the Development of Chronic Obstructive Pulmonary Disease through the Integration of Multiple Pathogenic Mechanisms. FASEB J. 2021, 35, e21376. [Google Scholar] [CrossRef]
- Jiang, L.; Fei, D.; Gong, R.; Yang, W.; Yu, W.; Pan, S.; Zhao, M.; Zhao, M. CORM-2 Inhibits TXNIP/NLRP3 Inflammasome Pathway in LPS-Induced Acute Lung Injury. Inflamm. Res. 2016, 65, 905–915. [Google Scholar] [CrossRef]
- Wang, X.; Qin, W.; Song, M.; Zhang, Y.; Sun, B. Exogenous Carbon Monoxide Inhibits Neutrophil Infiltration in LPS-Induced Sepsis by Interfering with FPR1 via P38 MAPK but Not GRK2. Oncotarget 2016, 7, 34250–34265. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-C.; Li, J.-C.; Wang, S.-M.; Cheng, C.-H.; Yeh, C.-H.; Lin, L.-S.; Chiu, H.-Y.; Chang, C.-Y.; Chuu, J.-J. The Effects of Carbon Monoxide Releasing Molecules on Paraquat-Induced Pulmonary Interstitial Inflammation and Fibrosis. Toxicology 2021, 456, 152750. [Google Scholar] [CrossRef]
- Lin, H.; Wang, X. The Effects of Gasotransmitters on Bronchopulmonary Dysplasia. Eur. J. Pharmacol. 2020, 873, 172983. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-W.; Wu, C.-H.; Chiang, Y.-C.; Chen, Y.-L.; Chang, K.-T.; Chuang, C.-C.; Lee, I.-T. Carbon Monoxide Releasing Molecule-2 Attenuates Pseudomonas Aeruginosa-Induced ROS-Dependent ICAM-1 Expression in Human Pulmonary Alveolar Epithelial Cells. Redox Biol. 2018, 18, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Tsoyi, K.; Hall, S.R.R.; Dalli, J.; Colas, R.A.; Ghanta, S.; Ith, B.; Coronata, A.; Fredenburgh, L.E.; Baron, R.M.; Choi, A.M.K.; et al. Carbon Monoxide Improves Efficacy of Mesenchymal Stromal Cells During Sepsis by Production of Specialized Proresolving Lipid Mediators. Crit. Care Med. 2016, 44, e1236–e1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-W.; Chi, M.-C.; Hsu, L.-F.; Yang, C.-M.; Hsu, T.-H.; Chuang, C.-C.; Lin, W.-N.; Chu, P.-M.; Lee, I.-T. Carbon Monoxide Releasing Molecule-2 Protects against Particulate Matter-Induced Lung Inflammation by Inhibiting TLR2 and 4/ROS/NLRP3 Inflammasome Activation. Mol. Immunol. 2019, 112, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Zeglinski, M.R.; Turner, C.T.; Zeng, R.; Schwartz, C.; Santacruz, S.; Pawluk, M.A.; Zhao, H.; Chan, A.W.H.; Carlsten, C.; Granville, D.J. Soluble Wood Smoke Extract Promotes Barrier Dysfunction in Alveolar Epithelial Cells through a MAPK Signaling Pathway. Sci. Rep. 2019, 9, 10027. [Google Scholar] [CrossRef]
- Tatsuta, M.; Kan-O, K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of Cigarette Smoke on Barrier Function and Tight Junction Proteins in the Bronchial Epithelium: Protective Role of Cathelicidin LL-37. Respir. Res. 2019, 20, 251. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Y.; Xu, M.; Zhang, H.; Chen, Y.; Chung, K.F.; Adcock, I.M.; Li, F. Roles of TRPA1 and TRPV1 in Cigarette Smoke -Induced Airway Epithelial Cell Injury Model. Free Radic. Biol. Med. 2019, 134, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Larcombe, A.N.; Papini, M.G.; Chivers, E.K.; Berry, L.J.; Lucas, R.M.; Wyrwoll, C.S. Mouse Lung Structure and Function after Long-Term Exposure to an Atmospheric Carbon Dioxide Level Predicted by Climate Change Modeling. Environ. Health Perspect 2021, 129, 17001. [Google Scholar] [CrossRef]
- Schneberger, D.; Pandher, U.; Thompson, B.; Kirychuk, S. Effects of Elevated CO2 Levels on Lung Immune Response to Organic Dust and Lipopolysaccharide. Respir. Res. 2021, 22, 104. [Google Scholar] [CrossRef]
- Schneberger, D.; DeVasure, J.M.; Bailey, K.L.; Romberger, D.J.; Wyatt, T.A. Effect of Low-Level CO2 on Innate Inflammatory Protein Response to Organic Dust from Swine Confinement Barns. J. Occup Med. Toxicol. 2017, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Shigemura, M.; Lecuona, E.; Angulo, M.; Dada, L.A.; Edwards, M.B.; Welch, L.C.; Casalino-Matsuda, S.M.; Sporn, P.H.S.; Vadász, I.; Helenius, I.T.; et al. Elevated CO2 Regulates the Wnt Signaling Pathway in Mammals, Drosophila Melanogaster and Caenorhabditis Elegans. Sci. Rep. 2019, 9, 18251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemura, M.; Sznajder, J.I. Elevated CO2 Modulates Airway Contractility. Interface Focus 2021, 11, 20200021. [Google Scholar] [CrossRef]
- Bharat, A.; Angulo, M.; Sun, H.; Akbarpour, M.; Alberro, A.; Cheng, Y.; Shigemura, M.; Berdnikovs, S.; Welch, L.C.; Kanter, J.A.; et al. High CO2 Levels Impair Lung Wound Healing. Am. J. Respir. Cell Mol. Biol. 2020, 63, 244–254. [Google Scholar] [CrossRef]
- Bălă, G.-P.; Râjnoveanu, R.-M.; Tudorache, E.; Motișan, R.; Oancea, C. Air Pollution Exposure—The (in)Visible Risk Factor for Respiratory Diseases. Environ. Sci. Pollut Res. 2021. [Google Scholar] [CrossRef]
- Nurmatov, U.B.; Tagiyeva, N.; Semple, S.; Devereux, G.; Sheikh, A. Volatile Organic Compounds and Risk of Asthma and Allergy: A Systematic Review. Eur. Respir. Rev. 2015, 24, 92–101. [Google Scholar] [CrossRef]
- Junge, K.M.; Buchenauer, L.; Elter, E.; Butter, K.; Kohajda, T.; Herberth, G.; Röder, S.; Borte, M.; Kiess, W.; von Bergen, M.; et al. Wood Emissions and Asthma Development: Results from an Experimental Mouse Model and a Prospective Cohort Study. Environ. Int. 2021, 151, 106449. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.; Jang, Y.-J.; Kim, J.-W.; Park, M.-J.; Yoo, Y.-M.; Jeung, E.-B. Anti-Asthmatic Effects of Volatile Organic Compounds from Chamaecyparis Obtusa, Pinus Densiflora, Pinus Koraiensis, or Larix Kaempferi Wood Panels. J. Physiol. Pharm. 2018, 69. [Google Scholar] [CrossRef]
- Ma, T.; Wang, X.; Li, L.; Sun, B.; Zhu, Y.; Xia, T. Electronic Cigarette Aerosols Induce Oxidative Stress-Dependent Cell Death and NF-ΚB Mediated Acute Lung Inflammation in Mice. Arch. Toxicol. 2021, 95, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Mandler, W.K.; Qi, C.; Orandle, M.S.; Sarkisian, K.; Mercer, R.R.; Stefaniak, A.B.; Knepp, A.K.; Bowers, L.N.; Battelli, L.A.; Shaffer, J.; et al. Mouse Pulmonary Response to Dust from Sawing Corian®, a Solid-Surface Composite Material. J. Toxicol. Environ. Health A 2019, 82, 645–663. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-G.; Hwang, Y.-H.; Mun, S.-K.; Kim, S.-J.; Jang, H.-Y.; Kim, H.; Paik, M.-J.; Yee, S.-T. Role of Th2 Cytokines on the Onset of Asthma Induced by Meta-Xylene in Mice. Environ. Toxicol. 2019, 34, 1121–1128. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Liu, W.; Jin, Y.; Guo, L. Effects of Subchronic Exposure to Low-Dose Volatile Organic Compounds on Lung Inflammation in Mice. Environ. Toxicol. 2014, 29, 1089–1097. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Liu, W.; Jin, Y. Oxidative Damage and Genotoxic Effect in Mice Caused by Sub-Chronic Exposure to Low-Dose Volatile Organic Compounds. Inhal. Toxicol. 2013, 25, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Im, U.; Brandt, J.; Geels, C.; Hansen, K.M.; Christensen, J.H.; Andersen, M.S.; Solazzo, E.; Kioutsioukis, I.; Alyuz, U.; Balzarini, A.; et al. Assessment and Economic Valuation of Air Pollution Impacts on Human Health over Europe and the United States as Calculated by a Multi-Model Ensemble in the Framework of AQMEII3. Atmos. Chem. Phys. 2018, 18, 5967–5989. [Google Scholar] [CrossRef] [Green Version]
- Gostner, J.M.; Zeisler, J.; Alam, M.T.; Gruber, P.; Fuchs, D.; Becker, K.; Neubert, K.; Kleinhappl, M.; Martini, S.; Überall, F. Cellular Reactions to Long-Term Volatile Organic Compound (VOC) Exposures. Sci. Rep. 2016, 6, 37842. [Google Scholar] [CrossRef] [PubMed]
- Alford, K.L.; Kumar, N. Pulmonary Health Effects of Indoor Volatile Organic Compounds-A Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 1578. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.-W.; Park, H.-W.; Kim, W.J.; Kim, M.-G.; Lee, S.-J. Exposure to Volatile Organic Compounds and Airway Inflammation. Environ. Health 2018, 17, 65. [Google Scholar] [CrossRef]
- Azim, A.; Barber, C.; Dennison, P.; Riley, J.; Howarth, P. Exhaled Volatile Organic Compounds in Adult Asthma: A Systematic Review. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Neerincx, A.H.; Vijverberg, S.J.H.; Bos, L.D.J.; Brinkman, P.; van der Schee, M.P.; de Vries, R.; Sterk, P.J.; der Zee, A.-H.M. Breathomics from Exhaled Volatile Organic Compounds in Pediatric Asthma. Pediatric. Pulmonol. 2017, 52, 1616–1627. [Google Scholar] [CrossRef]
- Choi, H.; Harrison, R.; Komulainen, H.; Saborit, J.M.D. Polycyclic Aromatic Hydrocarbons; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Sun, Y.; Shi, Z.; Lin, Y.; Zhang, M.; Liu, J.; Zhu, L.; Chen, Q.; Bi, J.; Li, S.; Ni, Z.; et al. Benzo(a)Pyrene Induces MUC5AC Expression through the AhR/Mitochondrial ROS/ERK Pathway in Airway Epithelial Cells. Ecotoxicol. Environ. Saf. 2021, 210, 111857. [Google Scholar] [CrossRef]
- Yanagisawa, R.; Koike, E.; Win-Shwe, T.-T.; Ichinose, T.; Takano, H. Effects of Lactational Exposure to Low-Dose BaP on Allergic and Non-Allergic Immune Responses in Mice Offspring. J. Immunotoxicol. 2018, 15, 31–40. [Google Scholar] [CrossRef]
- Yanagisawa, R.; Koike, E.; Win-Shwe, T.-T.; Ichinose, T.; Takano, H. Low-Dose Benzo[a]Pyrene Aggravates Allergic Airway Inflammation in Mice. J. Appl. Toxicol. 2016, 36, 1496–1504. [Google Scholar] [CrossRef]
- Wang, E.; Liu, X.; Tu, W.; Do, D.C.; Yu, H.; Yang, L.; Zhou, Y.; Xu, D.; Huang, S.-K.; Yang, P.; et al. Benzo(a)Pyrene Facilitates Dermatophagoides Group 1 (Der f 1)-Induced Epithelial Cytokine Release through Aryl Hydrocarbon Receptor in Asthma. Allergy 2019, 74, 1675–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, P.H.; Kitamura, G.; Honda, A.; Sawahara, T.; Hayashi, T.; Fukushima, W.; Kudo, H.; Ito, S.; Yoshida, S.; Ichinose, T.; et al. Synergistic Effect of Carbon Nuclei and Polyaromatic Hydrocarbons on Respiratory and Immune Responses. Environ. Toxicol. 2017, 32, 2172–2181. [Google Scholar] [CrossRef] [Green Version]
- Gdula-Argasińska, J.; Czepiel, J.; Totoń-Żurańska, J.; Wołkow, P.; Librowski, T.; Czapkiewicz, A.; Perucki, W.; Woźniakiewicz, M.; Woźniakiewicz, A. N-3 Fatty Acids Regulate the Inflammatory-State Related Genes in the Lung Epithelial Cells Exposed to Polycyclic Aromatic Hydrocarbons. Pharm. Rep. 2016, 68, 319–328. [Google Scholar] [CrossRef]
- Li, F.; Xiang, B.; Jin, Y.; Li, C.; Li, J.; Ren, S.; Huang, H.; Luo, Q. Dysregulation of Lipid Metabolism Induced by Airway Exposure to Polycyclic Aromatic Hydrocarbons in C57BL/6 Mice. Environ. Pollut. 2019, 245, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-Y.; Kim, Y.-H.; Kang, M.-K.; Lee, E.-J.; Kim, D.-Y.; Oh, H.; Kim, S.-I.; Na, W.; Kang, I.-J.; Kang, Y.-H. Aesculetin Inhibits Airway Thickening and Mucus Overproduction Induced by Urban Particulate Matter through Blocking Inflammation and Oxidative Stress Involving TLR4 and EGFR. Antioxid 2021, 10, 494. [Google Scholar] [CrossRef]
- Li, P.; Wang, J.; Guo, F.; Zheng, B.; Zhang, X. A Novel Inhibitory Role of MicroRNA-224 in Particulate Matter 2.5-Induced Asthmatic Mice by Inhibiting TLR2. J. Cell Mol. Med. 2020, 24, 3040–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Liu, Y.; Guo, D.; He, W.; Zhao, L.; Xia, S. PM2.5-Induced Pulmonary Inflammation via Activating of the NLRP3/Caspase-1 Signaling Pathway. Environ. Toxicol. 2021, 36, 298–307. [Google Scholar] [CrossRef]
- Dai, M.-Y.; Chen, F.-F.; Wang, Y.; Wang, M.-Z.; Lv, Y.-X.; Liu, R.-Y. Particulate Matters Induce Acute Exacerbation of Allergic Airway Inflammation via the TLR2/NF-ΚB/NLRP3 Signaling Pathway. Toxicol. Lett. 2020, 321, 146–154. [Google Scholar] [CrossRef]
- Park, S.K.; Yeon, S.H.; Choi, M.-R.; Choi, S.H.; Lee, S.B.; Rha, K.; Kim, Y.M. Urban Particulate Matters May Affect Endoplasmic Reticulum Stress and Tight Junction Disruption in Nasal Epithelial Cells. Am. J. Rhinol. Allergy 2021, 19458924211004010. [Google Scholar] [CrossRef]
- Caraballo, J.C.; Yshii, C.; Westphal, W.; Moninger, T.; Comellas, A.P. Ambient Particulate Matter Affects Occludin Distribution and Increases Alveolar Transepithelial Electrical Conductance. Respirology 2011, 16, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-G.; Lee, S.-H.; Hong, J.; Lee, P.-H.; Jang, A.-S. Titanium Dioxide Particles Modulate Epithelial Barrier Protein, Claudin 7 in Asthma. Mol. Immunol. 2021, 132, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xu, Y.; Huang, L.; Wang, K.; Shen, H.; Li, Z. Pollution Characteristics and Toxic Effects of PM1.0 and PM2.5 in Harbin, China. Environ. Sci. Pollut Res. Int. 2021, 28, 13229–13242. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kwon, D.-I.; Kim, M.; Im, S.-H.; Lee, Y.J. Commensal Microbiome Expands Tγδ17 Cells in the Lung and Promotes Particulate Matter-Induced Acute Neutrophilia. Front. Immunol. 2021, 12, 645741. [Google Scholar] [CrossRef]
- Xu, M.; Wang, X.; Xu, L.; Zhang, H.; Li, C.; Liu, Q.; Chen, Y.; Chung, K.F.; Adcock, I.M.; Li, F. Chronic Lung Inflammation and Pulmonary Fibrosis after Multiple Intranasal Instillation of PM2.5 in Mice. Environ. Toxicol. 2021. [Google Scholar] [CrossRef]
- Rahmani, H.; Sadeghi, S.; Taghipour, N.; Roshani, M.; Amani, D.; Ghazanfari, T.; Mosaffa, N. The Effects of Particulate Matter on C57BL/6 Peritoneal and Alveolar Macrophages. Iran. J. Allergy Asthma Immunol. 2020, 19, 647–659. [Google Scholar] [CrossRef]
- Estrella, B.; Naumova, E.N.; Cepeda, M.; Voortman, T.; Katsikis, P.D.; Drexhage, H.A. Effects of Air Pollution on Lung Innate Lymphoid Cells: Review of In Vitro and In Vivo Experimental Studies. Int. J. Environ. Res. Public Health 2019, 16, 2347. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Fan, X.; Gu, W.; Ci, X.; Peng, L. Hyperoside Relieves Particulate Matter-Induced Lung Injury by Inhibiting AMPK/MTOR-Mediated Autophagy Deregulation. Pharm. Res. 2021, 167, 105561. [Google Scholar] [CrossRef]
- Li, H.-H.; Liu, C.-C.; Hsu, T.-W.; Lin, J.-H.; Hsu, J.-W.; Li, A.F.-Y.; Yeh, Y.-C.; Hung, S.-C.; Hsu, H.-S. Upregulation of ACE2 and TMPRSS2 by Particulate Matter and Idiopathic Pulmonary Fibrosis: A Potential Role in Severe COVID-19. Part Fibre Toxicol. 2021, 18, 11. [Google Scholar] [CrossRef]
- de Oliveira Alves, N.; Martins Pereira, G.; Di Domenico, M.; Costanzo, G.; Benevenuto, S.; de Oliveira Fonoff, A.M.; de Souza Xavier Costa, N.; Ribeiro Júnior, G.; Satoru Kajitani, G.; Cestari Moreno, N.; et al. Inflammation Response, Oxidative Stress and DNA Damage Caused by Urban Air Pollution Exposure Increase in the Lack of DNA Repair XPC Protein. Environ. Int. 2020, 145, 106150. [Google Scholar] [CrossRef]
- Pang, L.; Yu, P.; Liu, X.; Fan, Y.; Shi, Y.; Zou, S. Fine Particulate Matter Induces Airway Inflammation by Disturbing the Balance between Th1/Th2 and Regulation of GATA3 and Runx3 Expression in BALB/c Mice. Mol. Med. Rep. 2021, 23. [Google Scholar] [CrossRef]
- Grove, K.C.D.; Provoost, S.; Brusselle, G.G.; Joos, G.F.; Maes, T. Insights in Particulate Matter-induced Allergic Airway Inflammation: Focus on the Epithelium. Clin. Exp. Allergy 2018, 48, 773–786. [Google Scholar] [CrossRef]
- Kim, M.H.; Park, S.J.; Yang, W.M. Inhalation of Essential Oil from Mentha Piperita Ameliorates PM10-Exposed Asthma by Targeting IL-6/JAK2/STAT3 Pathway Based on a Network Pharmacological Analysis. Pharmaceuticals 2020, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, X.; An, X.; Zhang, L.; Li, X.; Wang, L.; Zhu, G. Continuous Exposure of PM2.5 Exacerbates Ovalbumin-Induced Asthma in Mouse Lung via a JAK-STAT6 Signaling Pathway. Adv. Clin. Exp. Med. 2020, 29, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Ran, Z.; An, Y.; Zhou, J.; Yang, J.; Zhang, Y.; Yang, J.; Wang, L.; Li, X.; Lu, D.; Zhong, J.; et al. Subchronic Exposure to Concentrated Ambient PM2.5 Perturbs Gut and Lung Microbiota as Well as Metabolic Profiles in Mice. Environ. Pollut. 2021, 272, 115987. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Li, S.-W.; Lin, C.-D.; Huang, M.-Z.; Lin, H.-J.; Chin, C.-Y.; Lai, Y.-R.; Chiu, C.-H.; Yang, C.-Y.; Lai, C.-H. Fine Particulate Matter Exposure Alters Pulmonary Microbiota Composition and Aggravates Pneumococcus-Induced Lung Pathogenesis. Front. Cell Dev. Biol. 2020, 8, 570484. [Google Scholar] [CrossRef]
- Lin, C.-H.; Tseng, C.-Y.; Chao, M.-W. Administration of Lactobacillus Paracasei HB89 Mitigates PM2.5-Induced Enhancement of Inflammation and Allergic Airway Response in Murine Asthma Model. PLoS ONE 2020, 15, e0243062. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Kim, H.; Bae, C.; Kim, J.; Nam, B.; Lee, Y.; Kim, J.; Park, S.; Lee, J.; Sim, J. Lactobacillus HY2782 and Bifidobacterium HY8002 Decrease Airway Hyperresponsiveness Induced by Chronic PM2.5 Inhalation in Mice. J. Med. Food 2020, 23, 575–583. [Google Scholar] [CrossRef]
- Celebi Sözener, Z.; Cevhertas, L.; Nadeau, K.; Akdis, M.; Akdis, C.A. Environmental Factors in Epithelial Barrier Dysfunction. J. Allergy Clin. Immunol. 2020, 145, 1517–1528. [Google Scholar] [CrossRef]
- Xian, M.; Ma, S.; Wang, K.; Lou, H.; Wang, Y.; Zhang, L.; Wang, C.; Akdis, C.A. Particulate Matter 2.5 Causes Deficiency in Barrier Integrity in Human Nasal Epithelial Cells. Allergy Asthma Immunol. Res. 2020, 12, 56–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Guo, Z.; Zhang, R.; Deng, C.; Xu, J.; Dong, W.; Hong, Z.; Yu, H.; Situ, H.; Liu, C.; et al. Nasal Epithelial Barrier Disruption by Particulate Matter ≤2.5 Μm via Tight Junction Protein Degradation. J. Appl Toxicol. 2018, 38, 678–687. [Google Scholar] [CrossRef]
- Wang, C.; Cai, J.; Chen, R.; Shi, J.; Yang, C.; Li, H.; Lin, Z.; Meng, X.; Liu, C.; Niu, Y.; et al. Personal Exposure to Fine Particulate Matter, Lung Function and Serum Club Cell Secretory Protein (Clara). Environ. Pollut. 2017, 225, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Huang, J.; Wang, L.; Chen, C.; Yang, D.; Jin, M.; Bai, C.; Song, Y. Urban Particulate Matter Triggers Lung Inflammation via the ROS-MAPK-NF-ΚB Signaling Pathway. J. Thorac. Dis. 2017, 9, 4398–4412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Wang, J.; Li, J.; Jiang, N.; Zhang, R.; Yang, W.; Yao, W.; Wu, W. Oxidative Stress and Endocytosis Are Involved in Upregulation of Interleukin-8 Expression in Airway Cells Exposed to PM2.5. Environ. Toxicol. 2016, 31, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, G.; Gao, X.; Zhang, Y.; Fan, W.; Jiang, J.; An, Z.; Li, J.; Song, J.; Wu, W. Oxidative Stress-Mediated Epidermal Growth Factor Receptor Activation Regulates PM2.5-Induced over-Secretion of pro-Inflammatory Mediators from Human Bronchial Epithelial Cells. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129672. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-M.; Wang, Q.; Xing, W.-W.; Long, M.-H.; Fu, W.-L.; Xia, W.-R.; Jin, C.; Guo, N.; Xu, D.-Q.; Xu, D.-G. PM2.5 Induces Autophagy-Mediated Cell Death via NOS2 Signaling in Human Bronchial Epithelium Cells. Int. J. Biol. Sci. 2018, 14, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, H.-E. Diesel Exhaust Particles. Inhal. Toxicol. 2007, 19, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Veazey, J.; Eliseeva, S.; Chalupa, D.; Elder, A.; Georas, S.N. Diesel Exhaust Particle Exposure Reduces Expression of the Epithelial Tight Junction Protein Tricellulin. Part Fibre Toxicol. 2020, 17, 52. [Google Scholar] [CrossRef]
- An, Y.-F.; Geng, X.-R.; Mo, L.-H.; Liu, J.-Q.; Yang, L.-T.; Zhang, X.-W.; Liu, Z.-G.; Zhao, C.-Q.; Yang, P.-C. The 3-Methyl-4-Nitrophenol (PNMC) Compromises Airway Epithelial Barrier Function. Toxicology 2018, 395, 9–14. [Google Scholar] [CrossRef]
- Brandt, E.B.; Bolcas, P.E.; Ruff, B.P.; Hershey, G.K.K. IL33 Contributes to Diesel Pollution-mediated Increase in Experimental Asthma Severity. Allergy 2020, 75, 2254–2266. [Google Scholar] [CrossRef]
- De Grove, K.C.; Provoost, S.; Braun, H.; Blomme, E.E.; Teufelberger, A.R.; Krysko, O.; Beyaert, R.; Brusselle, G.G.; Joos, G.F.; Maes, T. IL-33 Signalling Contributes to Pollutant-Induced Allergic Airway Inflammation. Clin. Exp. Allergy 2018, 48, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- De Grove, K.C.; Provoost, S.; Hendriks, R.W.; McKenzie, A.N.J.; Seys, L.J.M.; Kumar, S.; Maes, T.; Brusselle, G.G.; Joos, G.F. Dysregulation of Type 2 Innate Lymphoid Cells and TH2 Cells Impairs Pollutant-Induced Allergic Airway Responses. J. Allergy Clin. Immunol. 2017, 139, 246–257.e4. [Google Scholar] [CrossRef] [Green Version]
- Daniel, S.; Phillippi, D.; Schneider, L.J.; Nguyen, K.N.; Mirpuri, J.; Lund, A.K. Exposure to Diesel Exhaust Particles Results in Altered Lung Microbial Profiles, Associated with Increased Reactive Oxygen Species/Reactive Nitrogen Species and Inflammation, in C57Bl/6 Wildtype Mice on a High-Fat Diet. Part Fibre Toxicol. 2021, 18, 3. [Google Scholar] [CrossRef]
- Uh, S.-T.; Koo, S.M.; Kim, Y.; Kim, K.; Park, S.; Jang, A.S.; Kim, D.; Kim, Y.H.; Park, C.-S. The Activation of NLRP3-Inflammsome by Stimulation of Diesel Exhaust Particles in Lung Tissues from Emphysema Model and RAW 264.7 Cell Line. Korean J. Intern. Med. 2017, 32, 865–874. [Google Scholar] [CrossRef]
- Kumar, S.; Joos, G.; Boon, L.; Tournoy, K.; Provoost, S.; Maes, T. Role of Tumor Necrosis Factor-α and Its Receptors in Diesel Exhaust Particle-Induced Pulmonary Inflammation. Sci. Rep. 2017, 7, 11508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaesteker, T.; Vanhoffelen, E.; Trekels, K.; Jonckheere, A.-C.; Cremer, J.; Vanstapel, A.; Dilissen, E.; Bullens, D.; Dupont, L.J.; Vanoirbeek, J.A. Differential Effects of Intense Exercise and Pollution on the Airways in a Murine Model. Part Fibre Toxicol. 2021, 18, 12. [Google Scholar] [CrossRef]
- de Homdedeu, M.; Cruz, M.J.; Sánchez-Díez, S.; Gómez-Ollés, S.; Ojanguren, I.; Ma, D.; Muñoz, X. Role of Diesel Exhaust Particles in the Induction of Allergic Asthma to Low Doses of Soybean. Environ. Res. 2020, 110337. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, G.; Bin, P.; Meng, T.; Niu, Y.; Yang, M.; Zhang, L.; Duan, H.; Yu, T.; Dai, Y.; et al. Time-Course Effects of Antioxidants and Phase II Enzymes on Diesel Exhaust Particles-Induced Oxidative Damage in the Mouse Lung. Toxicol. Appl. Pharm. 2019, 366, 25–34. [Google Scholar] [CrossRef]
- Gibbs, J.L.; Dallon, B.W.; Lewis, J.B.; Walton, C.M.; Arroyo, J.A.; Reynolds, P.R.; Bikman, B.T. Diesel Exhaust Particle Exposure Compromises Alveolar Macrophage Mitochondrial Bioenergetics. Int. J. Mol. Sci. 2019, 20, 5598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-J.; Shimizu, T.; Shinkai, Y.; Ihara, T.; Sugamata, M.; Kato, K.; Kobayashi, M.; Hirata, Y.; Inagaki, H.; Uzuki, M.; et al. Nrf2 Lowers the Risk of Lung Injury via Modulating the Airway Innate Immune Response Induced by Diesel Exhaust in Mice. Biomedicines 2020, 8, 443. [Google Scholar] [CrossRef]
- Cattani-Cavalieri, I.; da Maia Valença, H.; Moraes, J.A.; Brito-Gitirana, L.; Romana-Souza, B.; Schmidt, M.; Valença, S.S. Dimethyl Fumarate Attenuates Lung Inflammation and Oxidative Stress Induced by Chronic Exposure to Diesel Exhaust Particles in Mice. Int. J. Mol. Sci. 2020, 21, 9658. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, J.S.; Lee, H.J.; Jang, J.-H.; Kim, J.-H.; Sim, W.J.; Lim, Y.-B.; Jung, J.-W.; Lim, H.J. Age and Gender Effects on Genotoxicity in Diesel Exhaust Particles Exposed C57BL/6 Mice. Biomolecules 2021, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Deering-Rice, C.E.; Memon, T.; Lu, Z.; Romero, E.G.; Cox, J.; Taylor-Clark, T.; Veranth, J.M.; Reilly, C.A. Differential Activation of TRPA1 by Diesel Exhaust Particles: Relationships between Chemical Composition, Potency, and Lung Toxicity. Chem. Res. Toxicol. 2019, 32, 1040–1050. [Google Scholar] [CrossRef]
- Robinson, R.K.; Birrell, M.A.; Adcock, J.J.; Wortley, M.A.; Dubuis, E.D.; Chen, S.; McGilvery, C.M.; Hu, S.; Shaffer, M.S.P.; Bonvini, S.J.; et al. Mechanistic Link between Diesel Exhaust Particles and Respiratory Reflexes. J. Allergy Clin. Immunol. 2018, 141, 1074–1084.e9. [Google Scholar] [CrossRef] [Green Version]
- Akopian, A.N.; Fanick, E.R.; Brooks, E.G. TRP Channels and Traffic-Related Environmental Pollution-Induced Pulmonary Disease. Semin. Immunopathol. 2016, 38, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Agopyan, N.; Head, J.; Yu, S.; Simon, S.A. TRPV1 Receptors Mediate Particulate Matter-Induced Apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L563–L572. [Google Scholar] [CrossRef] [PubMed]
- Wooding, D.J.; Ryu, M.H.; Hüls, A.; Lee, A.D.; Lin, D.T.S.; Rider, C.F.; Yuen, A.C.Y.; Carlsten, C. Particle Depletion Does Not Remediate Acute Effects of Traffic-Related Air Pollution and Allergen. A Randomized, Double-Blind Crossover Study. Am. J. Respir. Crit. Care Med. 2019, 200, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Upadhyay, S.; Xiong, X.; Malmlöf, M.; Sandström, T.; Gerde, P.; Palmberg, L. Multi-Cellular Human Bronchial Models Exposed to Diesel Exhaust Particles: Assessment of Inflammation, Oxidative Stress and Macrophage Polarization. Part Fibre Toxicol. 2018, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarcone, M.C.; Duistermaat, E.; van Schadewijk, A.; Jedynska, A.; Hiemstra, P.S.; Kooter, I.M. Cellular Response of Mucociliary Differentiated Primary Bronchial Epithelial Cells to Diesel Exhaust. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L111–L123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozturk, A.B.; Bayraktar, R.; Gogebakan, B.; Mumbuc, S.; Bayram, H. Comparison of Inflammatory Cytokine Release from Nasal Epithelial Cells of Non-Atopic Non-Rhinitic, Allergic Rhinitic and Polyp Subjects and Effects of Diesel Exhaust Particles In Vitro. Allergol. Immunopathol. 2017, 45, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Savary, C.C.; Bellamri, N.; Morzadec, C.; Langouët, S.; Lecureur, V.; Vernhet, L. Long Term Exposure to Environmental Concentrations of Diesel Exhaust Particles Does Not Impact the Phenotype of Human Bronchial Epithelial Cells. Toxicol. Vitr. 2018, 52, 154–160. [Google Scholar] [CrossRef]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Wenzel, S.E. Asthma Phenotypes: The Evolution from Clinical to Molecular Approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Bontinck, A.; Maes, T.; Joos, G. Asthma and Air Pollution: Recent Insights in Pathogenesis and Clinical Implications. Curr. Opin. Pulm. Med. 2020, 26, 10–19. [Google Scholar] [CrossRef]
- Gras, D.; Martinez-Anton, A.; Bourdin, A.; Garulli, C.; de Senneville, L.; Vachier, I.; Vitte, J.; Chanez, P. Human Bronchial Epithelium Orchestrates Dendritic Cell Activation in Severe Asthma. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.-M.; Lee, M.-J.; He, J.-R.; Chao, M.-W.; Wang, C.-H.; Kuo, H.-P. Diesel Exhaust Particles Up-Regulate Interleukin-17A Expression via ROS/NF-ΚB in Airway Epithelium. Biochem. Pharm. 2018, 151, 1–8. [Google Scholar] [CrossRef]
- Huang, Y.; Lou, H.; Wang, C.; Zhang, L. Impact of Cigarette Smoke and IL-17A Activation on Asthmatic Patients with Chronic Rhinosinusitis. Rhinology 2019, 57, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Sava, F.; MacNutt, M.J.; Carlsten, C.R. Nasal Neurogenic Inflammation Markers Increase after Diesel Exhaust Inhalation in Individuals with Asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 759–760. [Google Scholar] [CrossRef]
- Mumby, S.; Chung, K.F.; Adcock, I.M. Transcriptional Effects of Ozone and Impact on Airway Inflammation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Bayram, H.; Rusznak, C.; Khair, O.A.; Sapsford, R.J.; Abdelaziz, M.M. Effect of Ozone and Nitrogen Dioxide on the Permeability of Bronchial Epithelial Cell Cultures of Non-Asthmatic and Asthmatic Subjects. Clin. Exp. Allergy 2002, 32, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, C.; Blomberg, A.; Pui, M.; Sandstrom, T.; Wong, S.W.; Alexis, N.; Hirota, J. Diesel Exhaust Augments Allergen-Induced Lower Airway Inflammation in Allergic Individuals: A Controlled Human Exposure Study. Thorax 2016, 71, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, A.; Hirota, J.A.; Hackett, T.L.; McNagny, K.M.; Wilson, S.J.; Carlsten, C. Morphometric Analysis of Inflammation in Bronchial Biopsies Following Exposure to Inhaled Diesel Exhaust and Allergen Challenge in Atopic Subjects. Part Fibre Toxicol. 2016, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Ryu, M.H.; Lau, K.S.-K.; Wooding, D.J.; Fan, S.; Sin, D.D.; Carlsten, C. Particle Depletion of Diesel Exhaust Restores Allergen-Induced Lung-Protective Surfactant Protein D in Human Lungs. Thorax 2020, 75, 640–647. [Google Scholar] [CrossRef]
- Jung, K.H.; Lovinsky-Desir, S.; Yan, B.; Torrone, D.; Lawrence, J.; Jezioro, J.R.; Perzanowski, M.; Perera, F.P.; Chillrud, S.N.; Miller, R.L. Effect of Personal Exposure to Black Carbon on Changes in Allergic Asthma Gene Methylation Measured 5 Days Later in Urban Children: Importance of Allergic Sensitization. Clin. Epigenetics 2017, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Eisner, M.D.; Anthonisen, N.; Coultas, D.; Kuenzli, N.; Perez-Padilla, R.; Postma, D.; Romieu, I.; Silverman, E.K.; Balmes, J.R. Committee on Nonsmoking COPD, Environmental and Occupational Health Assembly An Official American Thoracic Society Public Policy Statement: Novel Risk Factors and the Global Burden of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 182, 693–718. [Google Scholar] [CrossRef]
- Huang, X.; Mu, X.; Deng, L.; Fu, A.; Pu, E.; Tang, T.; Kong, X. The Etiologic Origins for Chronic Obstructive Pulmonary Disease. Int. J. Chron Obs. Pulmon. Dis. 2019, 14, 1139–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, K.; Brune, K.A.; Putcha, N.; Mandke, P.; O’Neal, W.K.; Shade, D.; Srivastava, V.; Wang, M.; Lam, H.; An, S.S.; et al. Cigarette Smoke Disrupts Monolayer Integrity by Altering Epithelial Cell-Cell Adhesion and Cortical Tension. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L581–L591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Jørgensen, J.T.; Ljungman, P.; Pershagen, G.; Bellander, T.; Leander, K.; Magnusson, P.K.E.; Rizzuto, D.; Hvidtfeldt, U.A.; Raaschou-Nielsen, O.; et al. Long-Term Exposure to Low-Level Air Pollution and Incidence of Chronic Obstructive Pulmonary Disease: The ELAPSE Project. Environ. Int. 2021, 146, 106267. [Google Scholar] [CrossRef]
- Duan, R.-R.; Hao, K.; Yang, T. Air Pollution and Chronic Obstructive Pulmonary Disease. Chronic. Dis. Transl. Med. 2020, 6, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Doiron, D.; de Hoogh, K.; Probst-Hensch, N.; Fortier, I.; Cai, Y.; De Matteis, S.; Hansell, A.L. Air Pollution, Lung Function and COPD: Results from the Population-Based UK Biobank Study. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Berend, N. Contribution of Air Pollution to COPD and Small Airway Dysfunction. Respirology 2016, 21, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, B.; Platel, A.; Antherieu, S.; Alleman, L.Y.; Hardy, E.M.; Perdrix, E.; Grova, N.; Riffault, V.; Appenzeller, B.M.; Happillon, M.; et al. Genetic and Epigenetic Alterations in Normal and Sensitive COPD-Diseased Human Bronchial Epithelial Cells Repeatedly Exposed to Air Pollution-Derived PM2.5. Environ. Pollut. 2017, 230, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, B.; Happillon, M.; Antherieu, S.; Hardy, E.M.; Alleman, L.Y.; Grova, N.; Perdrix, E.; Appenzeller, B.M.; Lo Guidice, J.-M.; Coddeville, P.; et al. Differential Responses of Healthy and Chronic Obstructive Pulmonary Diseased Human Bronchial Epithelial Cells Repeatedly Exposed to Air Pollution-Derived PM4. Environ. Pollut. 2016, 218, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Lo Guidice, J.-M.; Marchetti, P.; et al. Air Pollution-Derived PM2.5 Impairs Mitochondrial Function in Healthy and Chronic Obstructive Pulmonary Diseased Human Bronchial Epithelial Cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Li, F.; Ryffel, B.; Togbe, D.; Chung, K.F. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Wu, S.; Ni, Y.; Li, H.; Pan, L.; Yang, D.; Baccarelli, A.A.; Deng, F.; Chen, Y.; Shima, M.; Guo, X. Short-Term Exposure to High Ambient Air Pollution Increases Airway Inflammation and Respiratory Symptoms in Chronic Obstructive Pulmonary Disease Patients in Beijing, China. Environ. Int. 2016, 94, 76–82. [Google Scholar] [CrossRef]
- Zhao, J.; Li, M.; Wang, Z.; Chen, J.; Zhao, J.; Xu, Y.; Wei, X.; Wang, J.; Xie, J. Role of PM2.5 in the Development and Progression of COPD and Its Mechanisms. Respir. Res. 2019, 20, 120. [Google Scholar] [CrossRef] [Green Version]
- Hulina-Tomašković, A.; Rajković, M.G.; Jelić, D.; Bosnar, M.; Sladoljev, L.; Grubišić, T.Ž.; Rumora, L. Pro-Inflammatory Effects of Extracellular Hsp70 on NCI-H292 Human Bronchial Epithelial Cell Line. Int. J. Exp. Pathol. 2019, 100, 320–329. [Google Scholar] [CrossRef]
- Daigle, C.C.; Chalupa, D.C.; Gibb, F.R.; Morrow, P.E.; Oberdörster, G.; Utell, M.J.; Frampton, M.W. Ultrafine Particle Deposition in Humans during Rest and Exercise. Inhal. Toxicol. 2003, 15, 539–552. [Google Scholar] [CrossRef]
- Löndahl, J.; Massling, A.; Pagels, J.; Swietlicki, E.; Vaclavik, E.; Loft, S. Size-Resolved Respiratory-Tract Deposition of Fine and Ultrafine Hydrophobic and Hygroscopic Aerosol Particles during Rest and Exercise. Inhal. Toxicol. 2007, 19, 109–116. [Google Scholar] [CrossRef]
- Bonini, M.; Silvers, W. Exercise-Induced Bronchoconstriction: Background, Prevalence, and Sport Considerations. Immunol. Allergy Clin. N. Am. 2018, 38, 205–214. [Google Scholar] [CrossRef]
- Pagani, L.G.; Santos, J.M.B.; Foster, R.; Rossi, M.; Luna Junior, L.A.; Katekaru, C.M.; de Sá, M.C.; Jonckheere, A.-C.; Almeida, F.M.; Amaral, J.B.; et al. The Effect of Particulate Matter Exposure on the Inflammatory Airway Response of Street Runners and Sedentary People. Atmosphere 2020, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- De Santos, J.M.B.D.; Foster, R.; Jonckheere, A.-C.; Rossi, M.; Luna Junior, L.A.; Katekaru, C.M.; de Sá, M.C.; Pagani, L.G.; de Almeida, F.M.; do Amaral, J.B.; et al. Outdoor Endurance Training with Air Pollutant Exposure Versus Sedentary Lifestyle: A Comparison of Airway Immune Responses. Int. J. Environ. Res. Public Health 2019, 16, 4418. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante de Sá, M.; Nakagawa, N.K.; Saldiva de André, C.D.; Carvalho-Oliveira, R.; de Santana Carvalho, T.; Nicola, M.L.; de André, P.A.; Nascimento Saldiva, P.H.; Vaisberg, M. Aerobic Exercise in Polluted Urban Environments: Effects on Airway Defense Mechanisms in Young Healthy Amateur Runners. J. Breath Res. 2016, 10, 046018. [Google Scholar] [CrossRef]
- Laeremans, M.; Dons, E.; Avila-Palencia, I.; Carrasco-Turigas, G.; Orjuela, J.P.; Anaya, E.; Cole-Hunter, T.; de Nazelle, A.; Nieuwenhuijsen, M.; Standaert, A.; et al. Short-Term Effects of Physical Activity, Air Pollution and Their Interaction on the Cardiovascular and Respiratory System. Environ. Int. 2018, 117, 82–90. [Google Scholar] [CrossRef]
- Elshazly, F.A.; Abdelbasset, W.K.; Elnaggar, R.K.; Tantawy, S.A. Effects of Second-Hand Smoking on Lung Functions in Athlete and Non-Athlete School-Aged Children—Observational Study. Afr. Health Sci. 2020, 20, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Lovinsky-Desir, S.; Jung, K.H.; Jezioro, J.R.; Torrone, D.Z.; de Planell-Saguer, M.; Yan, B.; Perera, F.P.; Rundle, A.G.; Perzanowski, M.S.; Chillrud, S.N.; et al. Physical Activity, Black Carbon Exposure, and DNA Methylation in the FOXP3 Promoter. Clin. Epigenetics 2017, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Laeremans, M.; Dons, E.; Avila-Palencia, I.; Carrasco-Turigas, G.; Orjuela-Mendoza, J.P.; Anaya-Boig, E.; Cole-Hunter, T.; DE Nazelle, A.; Nieuwenhuijsen, M.; Standaert, A.; et al. Black Carbon Reduces the Beneficial Effect of Physical Activity on Lung Function. Med. Sci. Sports Exerc. 2018, 50, 1875–1881. [Google Scholar] [CrossRef] [Green Version]
- Cole, C.A.; Carlsten, C.; Koehle, M.; Brauer, M. Particulate Matter Exposure and Health Impacts of Urban Cyclists: A Randomized Crossover Study. Environ. Health 2018, 17, 78. [Google Scholar] [CrossRef] [Green Version]
- Giles, L.V.; Carlsten, C.; Koehle, M.S. The Pulmonary and Autonomic Effects of High-Intensity and Low-Intensity Exercise in Diesel Exhaust. Environ. Health 2018, 17, 87. [Google Scholar] [CrossRef] [Green Version]
- Rundell, K.W.; Smoliga, J.M.; Bougault, V. Exercise-Induced Bronchoconstriction and the Air We Breathe. Immunol. Allergy Clin. N. Am. 2018, 38, 183–204. [Google Scholar] [CrossRef]
- Qin, F.; Yang, Y.; Wang, S.; Dong, Y.; Xu, M.; Wang, Z.; Zhao, J. Exercise and Air Pollutants Exposure: A Systematic Review and Meta-Analysis. Life Sci. 2019, 218, 153–164. [Google Scholar] [CrossRef]
- Gomes, E.C.; Stone, V.; Florida-James, G. Impact of Heat and Pollution on Oxidative Stress and CC16 Secretion after 8 Km Run. Eur. J. Appl. Physiol. 2011, 111, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-Y.; Gilbreath, S.; Barakatt, E. Respiratory Outcomes of Ultrafine Particulate Matter (UFPM) as a Surrogate Measure of near-Roadway Exposures among Bicyclists. Environ. Health 2017, 16, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFlorio-Barker, S.; Lobdell, D.T.; Stone, S.L.; Boehmer, T.; Rappazzo, K.M. Acute Effects of Short-Term Exposure to Air Pollution While Being Physically Active, the Potential for Modification: A Review of the Literature. Prev. Med. 2020, 139, 106195. [Google Scholar] [CrossRef] [PubMed]
- Marmett, B.; Carvalho, R.B.; Dorneles, G.P.; Nunes, R.B.; Rhoden, C.R. Should I Stay or Should I Go: Can Air Pollution Reduce the Health Benefits of Physical Exercise? Med. Hypotheses 2020, 144, 109993. [Google Scholar] [CrossRef]
- Zhao, C.; Fang, X.; Feng, Y.; Fang, X.; He, J.; Pan, H. Emerging Role of Air Pollution and Meteorological Parameters in COVID-19. J. Evid. Based Med. 2021, 14, 123–138. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goossens, J.; Jonckheere, A.-C.; Dupont, L.J.; Bullens, D.M.A. Air Pollution and the Airways: Lessons from a Century of Human Urbanization. Atmosphere 2021, 12, 898. https://doi.org/10.3390/atmos12070898
Goossens J, Jonckheere A-C, Dupont LJ, Bullens DMA. Air Pollution and the Airways: Lessons from a Century of Human Urbanization. Atmosphere. 2021; 12(7):898. https://doi.org/10.3390/atmos12070898
Chicago/Turabian StyleGoossens, Janne, Anne-Charlotte Jonckheere, Lieven J. Dupont, and Dominique M. A. Bullens. 2021. "Air Pollution and the Airways: Lessons from a Century of Human Urbanization" Atmosphere 12, no. 7: 898. https://doi.org/10.3390/atmos12070898
APA StyleGoossens, J., Jonckheere, A.-C., Dupont, L. J., & Bullens, D. M. A. (2021). Air Pollution and the Airways: Lessons from a Century of Human Urbanization. Atmosphere, 12(7), 898. https://doi.org/10.3390/atmos12070898