
A Year-Round Measurement of Water-Soluble Trace and Rare Earth Elements in Arctic Aerosol: Possible Inorganic Tracers of Specific Events

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Instrumental Analysis
2.3. Enrichment Factor
2.4. Chemometric Approach and Back Trajectories Analysis
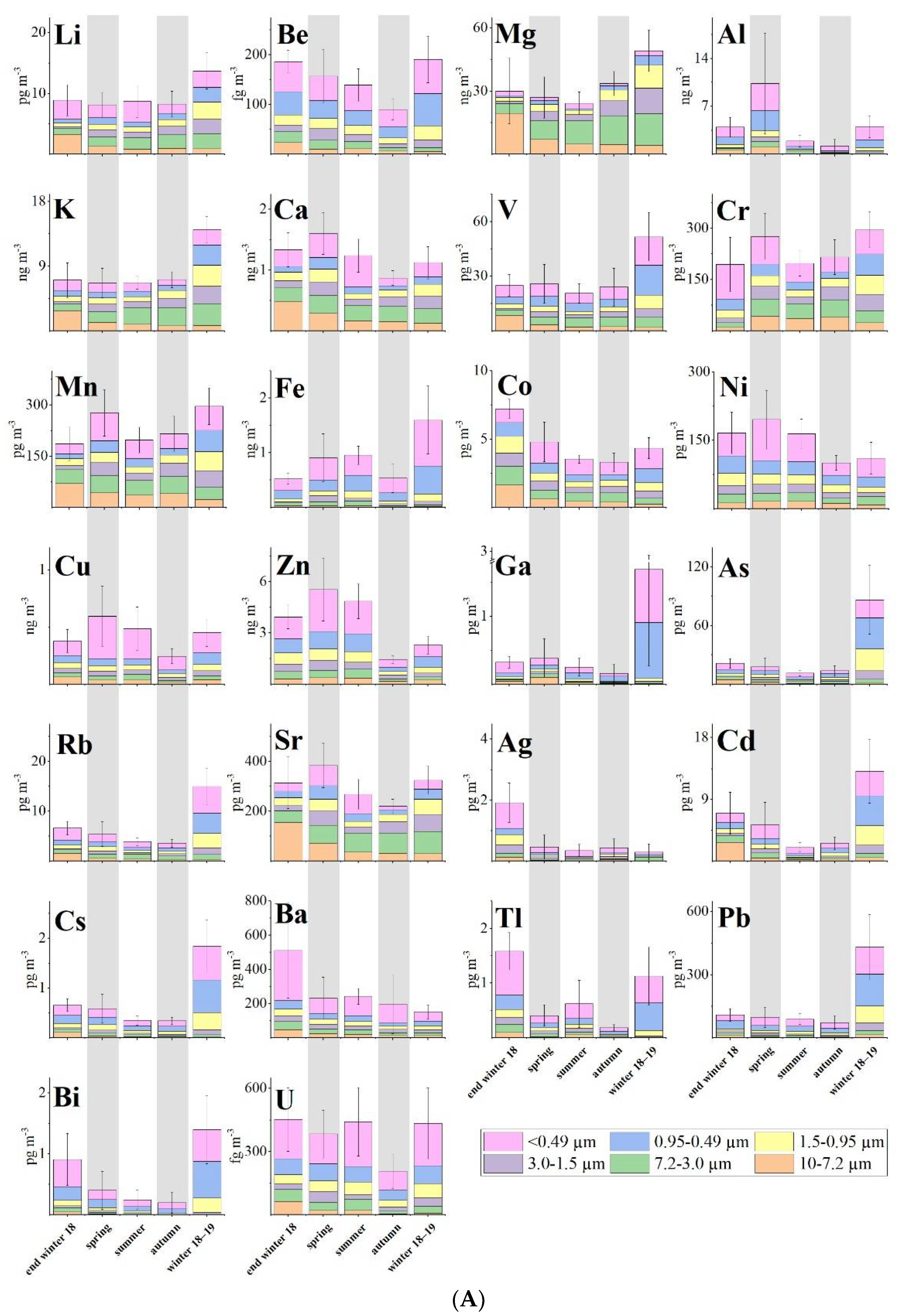
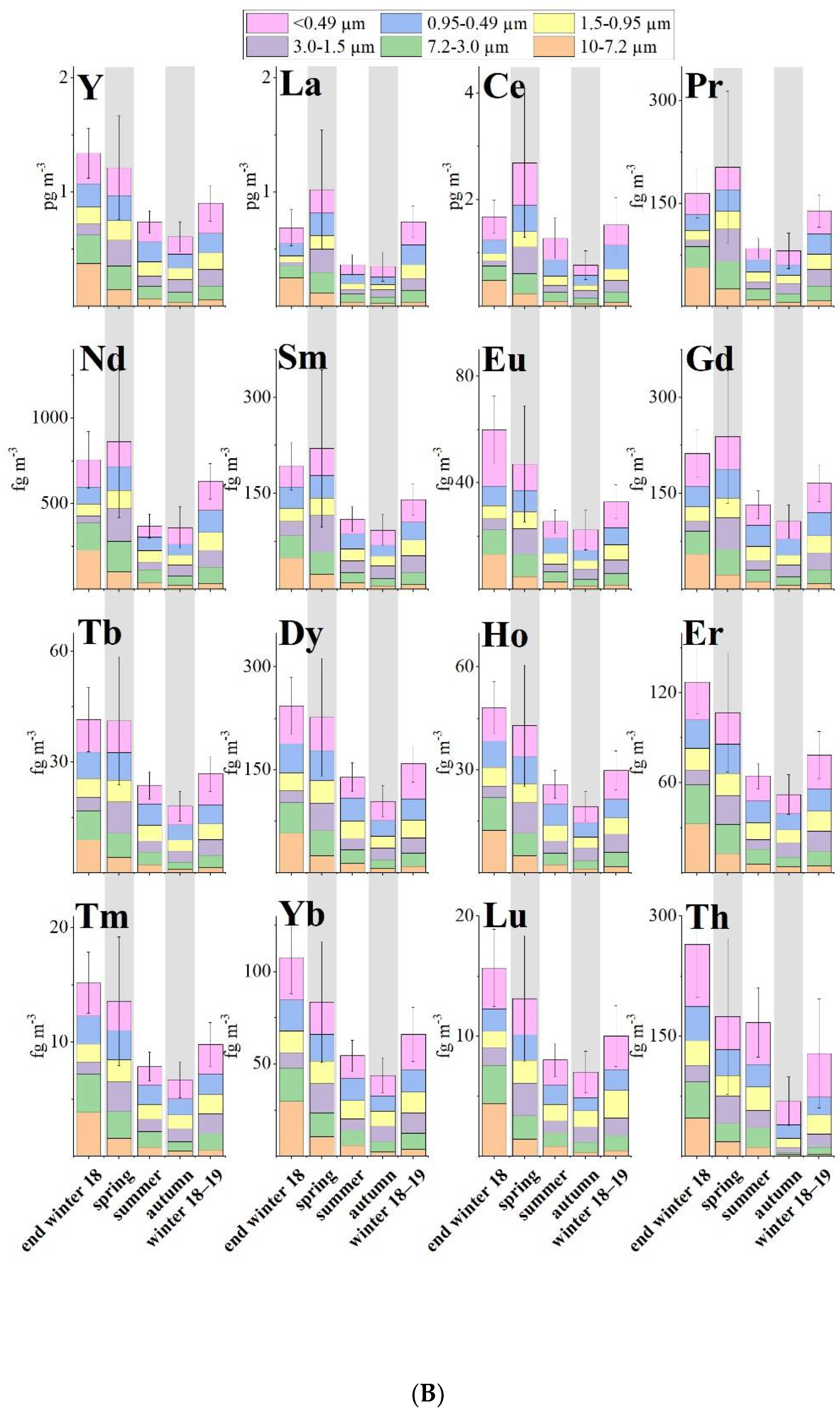
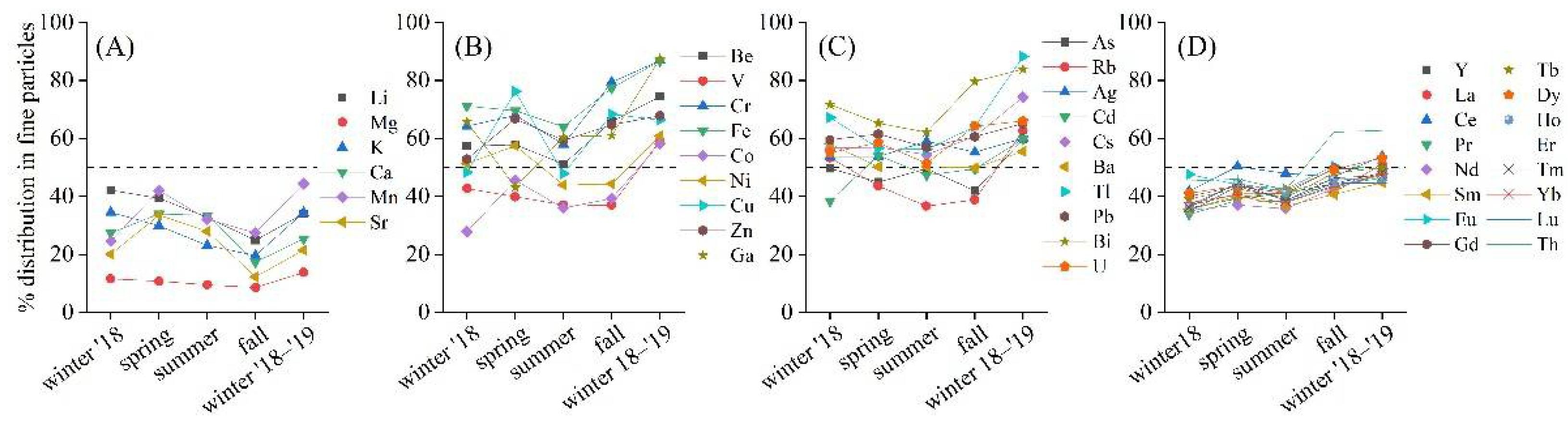
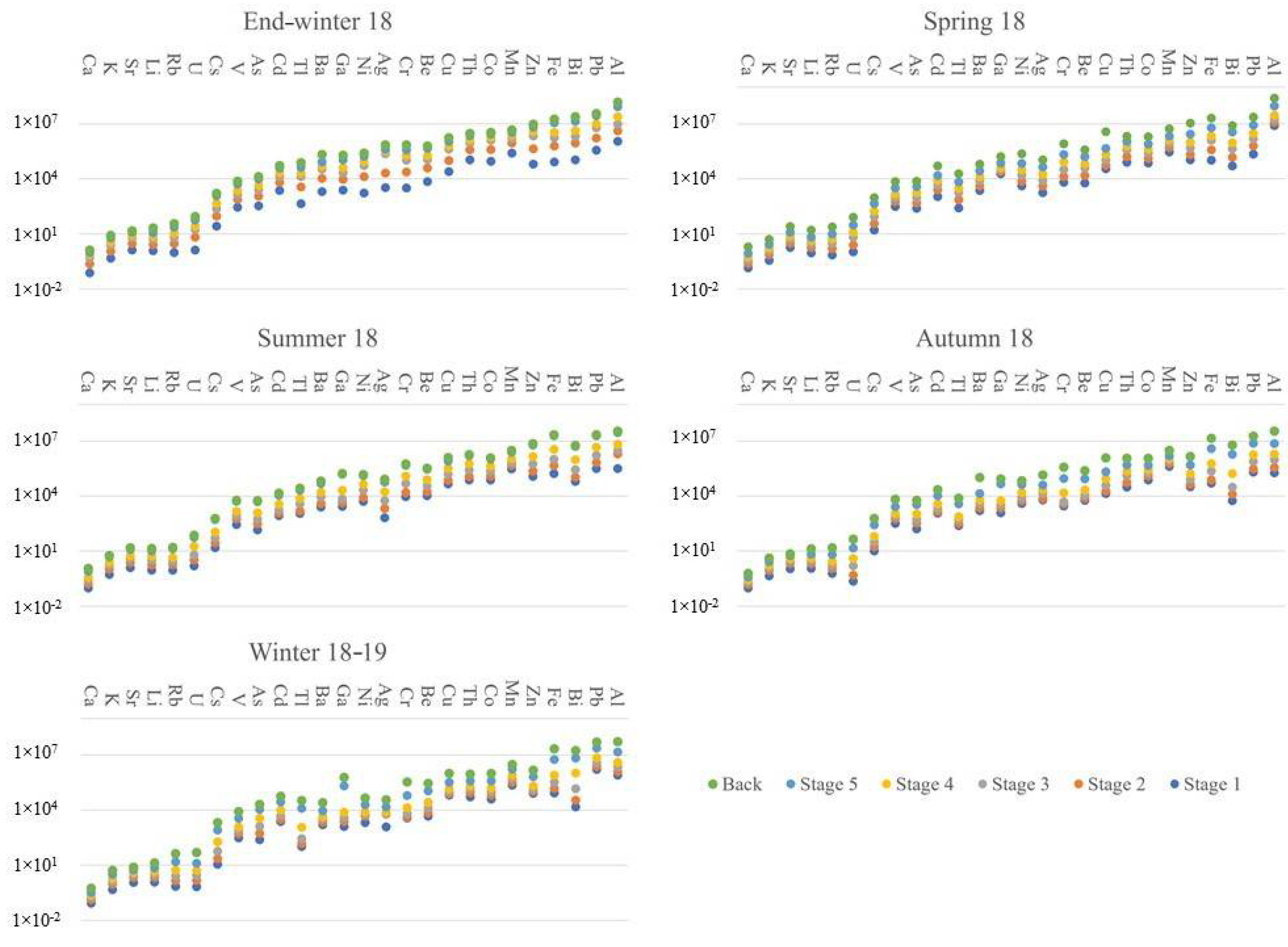
3. Results
4. Discussion
4.1. Enrichment Factors
4.2. Ternary Diagrams
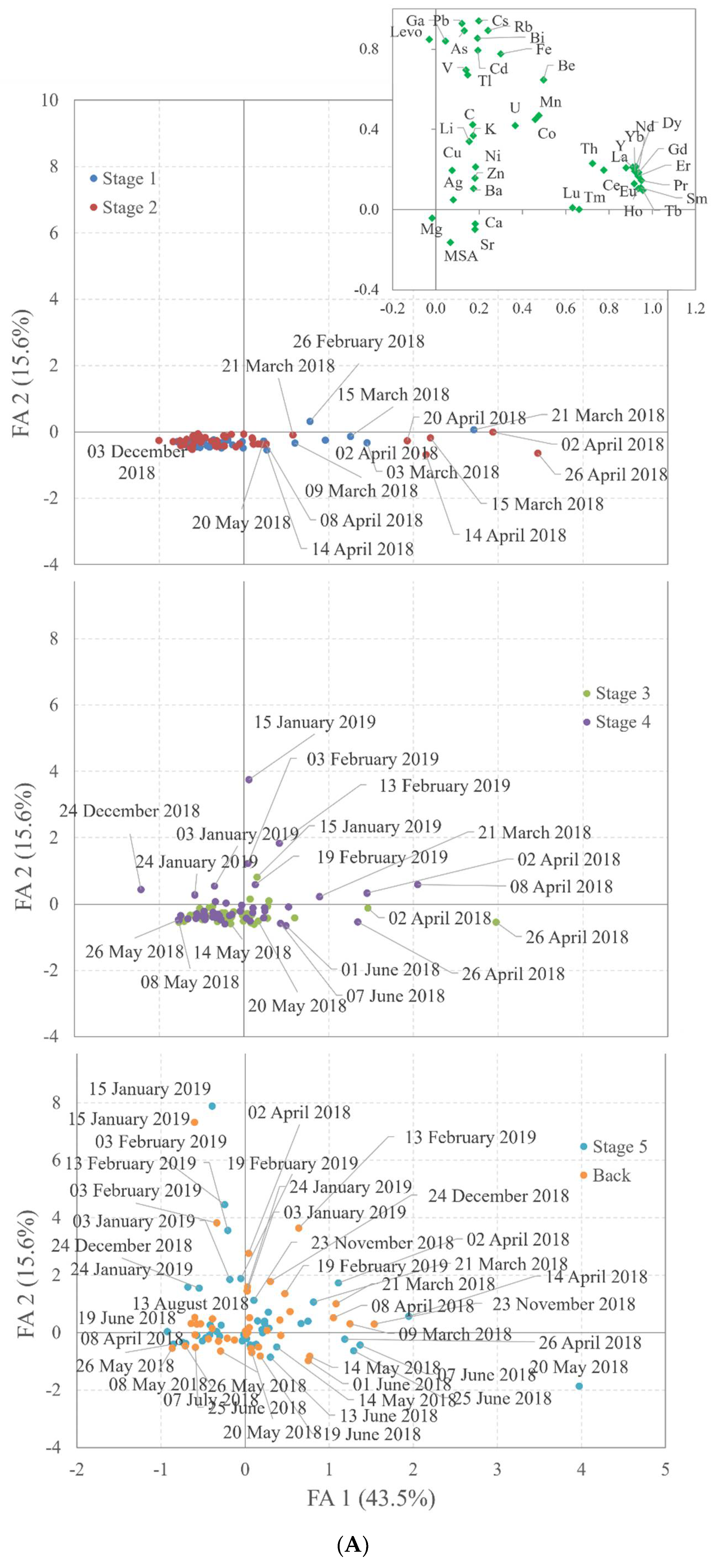
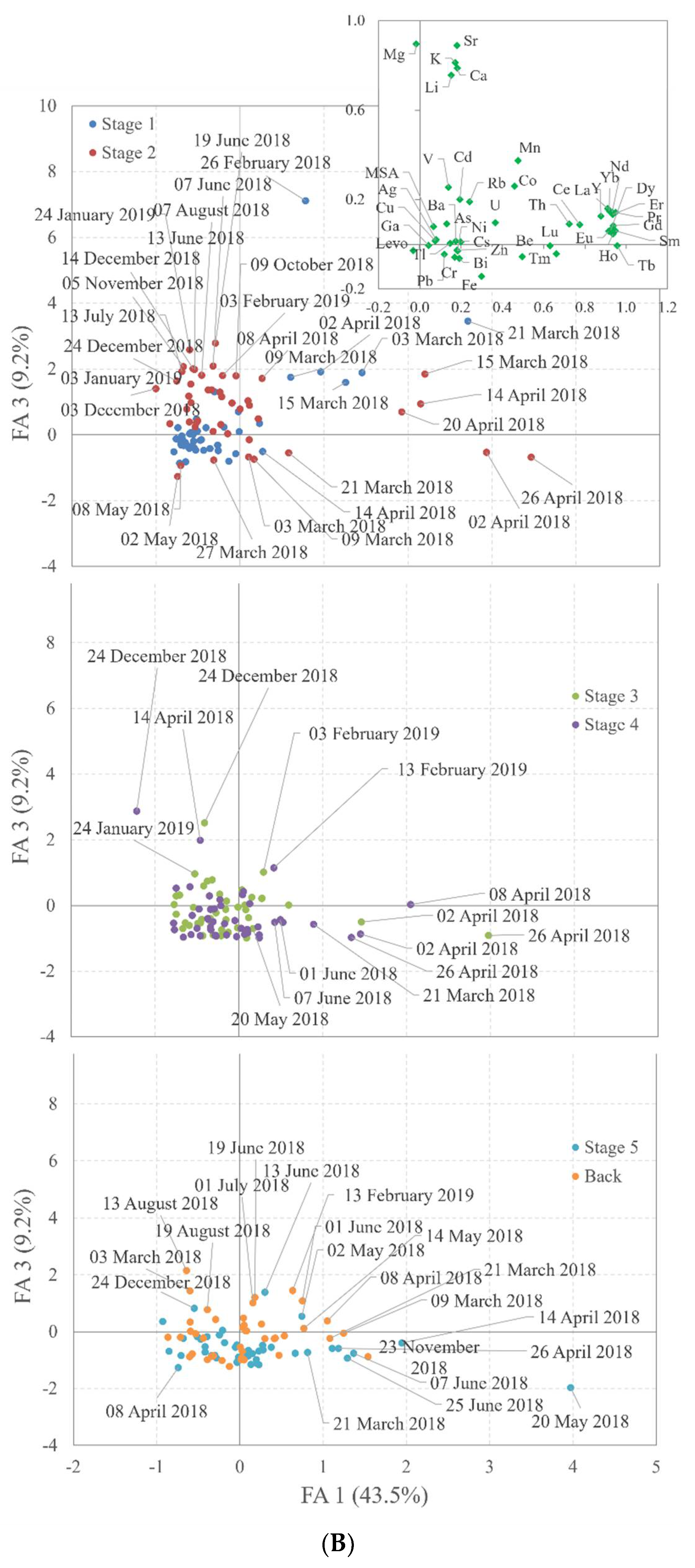
4.3. Chemometric Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maturilli, M.; Hanssen-Bauer, I.; Neuber, R.; Rex, M.; Edvardsen, K. The Atmosphere Above Ny-Ålesund: Climate and Global Warming, Ozone and Surface UV Radiation. In The Ecosystem of Kongsfjorden, Svalbard; Springer: Cham, Switzerland, 2019; pp. 23–46. ISBN 978-3-319-46425-1. [Google Scholar]
- Serreze, M.C.; Barry, R.G. Processes and impacts of Arctic amplification: A research synthesis. Glob. Planet. Chang. 2011, 77, 85–96. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; Wiley: New York, NY, USA, 2006; ISBN 9780471720171. [Google Scholar]
- Haywood, J.; Boucher, O. Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review. Rev. Geophys. 2000, 38, 513–543. [Google Scholar] [CrossRef]
- Landsberger, S. Multielemental composition of the Arctic aerosol. JGR Atmos. 1990, 95, 3509–3515. [Google Scholar] [CrossRef]
- Spolaor, A.; Moroni, B.; Luks, B.; Nawrot, A.; Roman, M.; Larose, C.; Stachnik, Ł.; Bruschi, F.; Kozioł, K.; Pawlak, F.; et al. Investigation on the Sources and Impact of Trace Elements in the Annual Snowpack and the Firn in the Hansbreen (Southwest Spitsbergen). Front. Earth Sci. 2021, 8, 1–10. [Google Scholar] [CrossRef]
- Giardi, F.; Becagli, S.; Traversi, R.; Frosini, D.; Severi, M.; Caiazzo, L.; Ancillotti, C.; Cappelletti, D.; Moroni, B.; Grotti, M.; et al. Size distribution and ion composition of aerosol collected at Ny-Ålesund in the spring-summer field campaign 2013. Rend. Lincei 2016, 27, 47–58. [Google Scholar] [CrossRef]
- Udisti, R.; Bazzano, A.; Becagli, S.; Bolzacchini, E.; Caiazzo, L.; Cappelletti, D.; Ferrero, L.; Frosini, D.; Giardi, F.; Grotti, M.; et al. Sulfate source apportionment in the Ny-Ålesund (Svalbard Islands) Arctic aerosol. Rend. Lincei 2016, 27, 85–94. [Google Scholar] [CrossRef]
- Becagli, S.; Amore, A.; Caiazzo, L.; Di Iorio, T.; Sarra, A.; Lazzara, L.; Marchese, C.; Meloni, D.; Mori, G.; Muscari, G.; et al. Biogenic Aerosol in the Artic from Eight Years of MSA Data from Ny Ålesund (Svalbard Islands) and Thule (Greenland). Atmosphere 2019, 10, 349. [Google Scholar] [CrossRef]
- Feltracco, M.; Barbaro, E.; Tedeschi, S.; Spolaor, A.; Turetta, C.; Vecchiato, M.; Morabito, E.; Zangrando, R.; Barbante, C.; Gambaro, A. Interannual variability of sugars in Arctic aerosol: Biomass burning and biogenic inputs. Sci. Total Environ. 2020, 706, 136089. [Google Scholar] [CrossRef] [PubMed]
- Zangrando, R.; Barbaro, E.; Zennaro, P.; Rossi, S.; Kehrwald, N.M.; Gabrieli, J.; Barbante, C.; Gambaro, A. Molecular markers of biomass burning in Arctic aerosols. Environ. Sci. Technol. 2013, 47, 8565–8574. [Google Scholar] [CrossRef]
- Feltracco, M.; Barbaro, E.; Hoppe, C.J.M.; Wolf, K.K.E.; Spolaor, A.; Layton, R.; Keuschnig, C.; Barbante, C.; Gambaro, A.; Larose, C. Airborne bacteria and particulate chemistry capture phytoplankton bloom dynamics in an Arctic Fjord. Under Rev. Atmos. Environ. 2021, 256, 118458. [Google Scholar] [CrossRef]
- Feltracco, M.; Barbaro, E.; Kirchgeorg, T.; Spolaor, A.; Turetta, C.; Zangrando, R.; Barbante, C.; Gambaro, A. Free and combined L- and D-amino acids in Arctic aerosol. Chemosphere 2019, 220, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Scalabrin, E.; Zangrando, R.; Barbaro, E.; Kehrwald, N.M.; Gabrieli, J.; Barbante, C.; Gambaro, A. Amino acids in Arctic aerosols. Atmos. Chem. Phys. 2012, 12, 10453–10463. [Google Scholar] [CrossRef]
- Stohl, A.; Berg, T.; Burkhart, J.F.; Fjæraa, A.M.; Forster, C.; Herber, A.; Hov, Ø.; Lunder, C.; McMillan, W.W.; Oltmans, S.; et al. Arctic smoke–record high air pollution levels in the European Arctic due to agricultural fires in Eastern Europe. Atmos. Chem. Phys. Discuss. 2006, 6, 9655–9722. [Google Scholar] [CrossRef]
- Eleftheriadis, K.; Vratolis, S.; Nyeki, S. Aerosol black carbon in the European Arctic: Measurements at Zeppelin station, Ny-Ålesund, Svalbard from 1998–2007. Geophys. Res. Lett. 2009, 36, 1–5. [Google Scholar] [CrossRef]
- Feltracco, M.; Barbaro, E.; Spolaor, A.; Vecchiato, M.; Callegaro, A.; Burgay, F.; Vardè, M.; Maffezzoli, N.; Dallo, F.; Scoto, F.; et al. Year-round measurements of size-segregated low molecular weight organic acids in Arctic aerosol. Sci. Total Environ. 2021, 763, 10. [Google Scholar] [CrossRef]
- Turetta, C.; Zangrando, R.; Barbaro, E.; Gabrieli, J.; Scalabrin, E.; Zennaro, P.; Gambaro, A.; Toscano, G.; Barbante, C. Water-soluble trace, rare earth elements and organic compounds in Arctic aerosol. Rend. Lincei 2016, 27, 95–103. [Google Scholar] [CrossRef]
- Conca, E.; Abollino, O.; Giacomino, A.; Buoso, S.; Traversi, R.; Becagli, S.; Grotti, M.; Malandrino, M. Source identification and temporal evolution of trace elements in PM10 collected near to Ny-Ålesund (Norwegian Arctic). Atmos. Environ. 2019, 203, 153–165. [Google Scholar] [CrossRef]
- Pacyna, J.M.; Ottar, B.; Tomza, U.; Maenhaut, W. Long-range transport of trace elements to Ny Ålesund, Spitsbergen. Atmos. Environ. 1985, 19, 857–865. [Google Scholar] [CrossRef]
- Giardi, F.; Traversi, R.; Becagli, S.; Severi, M.; Caiazzo, L.; Ancillotti, C.; Udisti, R. Determination of Rare Earth Elements in multi-year high-resolution Arctic aerosol record by double focusing Inductively Coupled Plasma Mass Spectrometry with desolvation nebulizer inlet system. Sci. Total Environ. 2018, 613–614, 1284–1294. [Google Scholar] [CrossRef]
- Quinn, P.K.; Shaw, G.; Andrews, E.; Dutton, E.G.; Ruoho-Airola, T.; Gong, S.L. Arctic haze: Current trends and knowledge gaps. Tellus Ser. B Chem. Phys. Meteorol. 2007, 59, 99–114. [Google Scholar] [CrossRef]
- Stohl, A. Characteristics of atmospheric transport into the Arctic troposphere. J. Geophys. Res. Atmos. 2006, 111, 1–17. [Google Scholar] [CrossRef]
- Toscano, G.; Gambaro, A.; Moret, I.; Capodaglio, G.; Turetta, C.; Cescon, P. Trace metals in aerosol at Terra Nova Bay, Antarctica. J. Environ. Monit. 2005, 7, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, E.; Kirchgeorg, T.; Zangrando, R.; Vecchiato, M.; Piazza, R.; Barbante, C.; Gambaro, A. Sugars in Antarctic aerosol. Atmos. Environ. 2015, 118, 135–144. [Google Scholar] [CrossRef]
- Barbaro, E.; Padoan, S.; Kirchgeorg, T.; Zangrando, R.; Toscano, G.; Barbante, C.; Gambaro, A. Particle size distribution of inorganic and organic ions in coastal and inland Antarctic aerosol. Environ. Sci. Pollut. Res. 2017, 24, 2724–2733. [Google Scholar] [CrossRef]
- Hans Wedepohl, K. The composition of the continental crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Gao, Y.; Arimoto, R.; Duce, R.A.; Lee, D.S.; Zhou, M.Y. Input of atmospheric trace elements and mineral matter to the Yellow Sea during the spring of a low-dust year. J. Geophys. Res. 1992, 97, 3767–3777. [Google Scholar] [CrossRef]
- Nozaki, Y. Elemental Distribution: Overview; Turekian, K.K., Ed.; A derivative of Encyclopedia of Ocean Sciences; Academic Press: Cambridge, MA, USA, 2010; ISBN 9780080964836. [Google Scholar]
- Basilevsky, A. Statistical Factor Analysis and Related Methods: Theory and Application; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1994; ISBN 0471570826. [Google Scholar]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. Noaa’s hysplit atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Draxler, R.R. An overview of the HYSPLIT_4 modelling system for trajectories, dispersion and deposition. Aust. Meteorol. Mag. 1998, 47, 295–308. [Google Scholar]
- Andreae, M.O. Aerosols before pollution. Science 2007, 315, 50–51. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Global Change; Wiley: Hoboken, NJ, USA, 1998; Volume 1360. [Google Scholar]
- Zhan, J.; Gao, Y.; Li, W.; Chen, L.; Lin, H.; Lin, Q. Effects of ship emissions on summertime aerosols at Ny-Alesund in the Arctic. Atmos. Pollut. Res. 2014, 5, 500–510. [Google Scholar] [CrossRef]
- Stohl, A.; Forster, C.; Eckhardt, S.; Spichtinger, N.; Huntrieser, H.; Heland, J.; Schlager, H.; Wilhelm, S.; Arnold, F.; Cooper, O. A backward modeling study of intercontinental pollution transport using aircraft measurements. J. Geophys. Res. Atmos. 2003, 108. [Google Scholar] [CrossRef]
- Simoneit, B.R.T.; Schauer, J.J.; Nolte, C.G.; Oros, D.R.; Elias, V.O.; Fraser, M.P.; Rogge, W.F.; Cass, G.R. Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles. Atmos. Environ. 1999, 33, 173–183. [Google Scholar] [CrossRef]
- Gondwe, M.; Krol, M.; Gieskes, W.; Klaassen, W.; de Baar, H. The contribution of ocean-leaving DMS to the global atmospheric burdens of DMS, MSA, SO2, and NSS SO4=. Glob. Biogeochem. Cycles 2003, 17. [Google Scholar] [CrossRef]
- Moreno, T.; Querol, X.; Alastuey, A.; Pey, J.; Minguillón, M.C.; Pérez, N.; Bernabé, R.M.; Blanco, S.; Cárdenas, B.; Gibbons, W. Lanthanoid geochemistry of urban atmospheric particulate matter. Environ. Sci. Technol. 2008, 42, 6502–6507. [Google Scholar] [CrossRef]
- Viana, M.; Kuhlbusch, T.A.J.; Querol, X.; Alastuey, A.; Harrison, R.M.; Hopke, P.K.; Winiwarter, W.; Vallius, M.; Szidat, S.; Prévôt, A.S.H.; et al. Source apportionment of particulate matter in Europe: A review of methods and results. J. Aerosol. Sci. 2008, 39, 827–849. [Google Scholar] [CrossRef]
- Quinn, P.K.; Miller, T.L.; Bates, T.S.; Ogren, J.A.; Andrews, E.; Shaw, G.E. A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska. J. Geophys. Res. Atmos. 2002, 107, 8–15. [Google Scholar] [CrossRef]
- Maturilli, M. Continuous Meteorological Observations at Station Ny-Ålesund (2011-08 et seq). Alfred Wegener Institute-Research Unit Potsdam, PANGAEA. 2020. Available online: https://doi.pangaea.de/10.1594/PANGAEA.914979 (accessed on 23 April 2021).
- Feltracco, M.; Barbaro, E.; Spolaor, A.; Vecchiato, M.; Callegaro, A.; Burgay, F.; Vardè, M.; Maffezzoli, N.; Dallo, F.; Scoto, F.; et al. Water soluble compounds in size-segregated Arctic aerosol at Gruvebadet, Ny-Ålesund in 2013, 2014, 2015 and 2018–2019. PANGAEA 2021. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | F1 | F2 | F3 | F4 | F1 | F2 | F3 | F4 | |
|---|---|---|---|---|---|---|---|---|---|
| MSA | 0.07 | −0.16 | 0.08 | 0.76 | Y | 0.91 | 0.21 | 0.16 | 0.16 |
| Levo | −0.03 | 0.85 | −0.03 | −0.04 | La | 0.88 | 0.21 | 0.12 | 0.16 |
| Be | 0.50 | 0.65 | −0.06 | 0.25 | Ce | 0.78 | 0.20 | 0.09 | 0.45 |
| V | 0.14 | 0.70 | 0.25 | 0.14 | Pr | 0.95 | 0.14 | 0.14 | 0.05 |
| Fe | 0.30 | 0.78 | −0.14 | 0.37 | Nd | 0.93 | 0.18 | 0.14 | 0.07 |
| Ga | 0.04 | 0.84 | −0.01 | 0.03 | Sm | 0.95 | 0.11 | 0.06 | 0.07 |
| As | 0.13 | 0.89 | 0.09 | −0.07 | Eu | 0.92 | 0.13 | 0.06 | 0.09 |
| Rb | 0.24 | 0.89 | 0.19 | 0.07 | Gd | 0.94 | 0.19 | 0.08 | 0.19 |
| Cd | 0.19 | 0.79 | 0.20 | 0.11 | Tb | 0.96 | 0.10 | −0.01 | 0.06 |
| Cs | 0.20 | 0.94 | 0.01 | 0.05 | Dy | 0.92 | 0.21 | 0.06 | 0.19 |
| Tl | 0.15 | 0.67 | 0.00 | 0.17 | Ho | 0.94 | 0.10 | 0.05 | 0.08 |
| Pb | 0.12 | 0.93 | −0.04 | 0.11 | Er | 0.93 | 0.17 | 0.13 | 0.10 |
| Bi | 0.19 | 0.85 | −0.06 | 0.07 | Tm | 0.66 | 0.00 | −0.04 | −0.08 |
| Li | 0.15 | 0.34 | 0.76 | 0.01 | Yb | 0.92 | 0.19 | 0.15 | 0.07 |
| Mg | −0.02 | −0.04 | 0.90 | −0.31 | Lu | 0.63 | 0.01 | −0.01 | −0.06 |
| K | 0.17 | 0.37 | 0.81 | −0.16 | Th | 0.72 | 0.23 | 0.09 | 0.12 |
| Ca | 0.18 | −0.07 | 0.79 | 0.28 | Mn | 0.48 | 0.47 | 0.37 | 0.18 |
| Sr | 0.18 | −0.10 | 0.89 | 0.17 | Co | 0.46 | 0.45 | 0.26 | 0.36 |
| Cr | 0.17 | 0.42 | −0.06 | 0.64 | Ag | 0.08 | 0.05 | 0.02 | 0.21 |
| Ni | 0.18 | 0.21 | −0.03 | 0.71 | Ba | 0.17 | 0.11 | 0.01 | 0.19 |
| Cu | 0.08 | 0.20 | 0.02 | 0.85 | U | 0.37 | 0.42 | 0.10 | 0.38 |
| Zn | 0.18 | 0.16 | −0.03 | 0.83 | Expl. % | 43.5 | 59.1 | 68.3 | 75.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turetta, C.; Feltracco, M.; Barbaro, E.; Spolaor, A.; Barbante, C.; Gambaro, A. A Year-Round Measurement of Water-Soluble Trace and Rare Earth Elements in Arctic Aerosol: Possible Inorganic Tracers of Specific Events. Atmosphere 2021, 12, 694. https://doi.org/10.3390/atmos12060694
Turetta C, Feltracco M, Barbaro E, Spolaor A, Barbante C, Gambaro A. A Year-Round Measurement of Water-Soluble Trace and Rare Earth Elements in Arctic Aerosol: Possible Inorganic Tracers of Specific Events. Atmosphere. 2021; 12(6):694. https://doi.org/10.3390/atmos12060694
Chicago/Turabian StyleTuretta, Clara, Matteo Feltracco, Elena Barbaro, Andrea Spolaor, Carlo Barbante, and Andrea Gambaro. 2021. "A Year-Round Measurement of Water-Soluble Trace and Rare Earth Elements in Arctic Aerosol: Possible Inorganic Tracers of Specific Events" Atmosphere 12, no. 6: 694. https://doi.org/10.3390/atmos12060694
APA StyleTuretta, C., Feltracco, M., Barbaro, E., Spolaor, A., Barbante, C., & Gambaro, A. (2021). A Year-Round Measurement of Water-Soluble Trace and Rare Earth Elements in Arctic Aerosol: Possible Inorganic Tracers of Specific Events. Atmosphere, 12(6), 694. https://doi.org/10.3390/atmos12060694