
Abundance of NO3 Derived Organo-Nitrates and Their Importance in the Atmosphere


 , ,
, ,  and
and 
Abstract
:1. Introduction
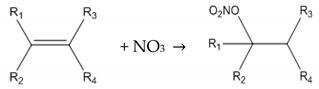
2. Methodology
2.1. Measurement Site and Measurement Technique
2.2. WRF-Chem-CRI Model
3. Results and Discussion
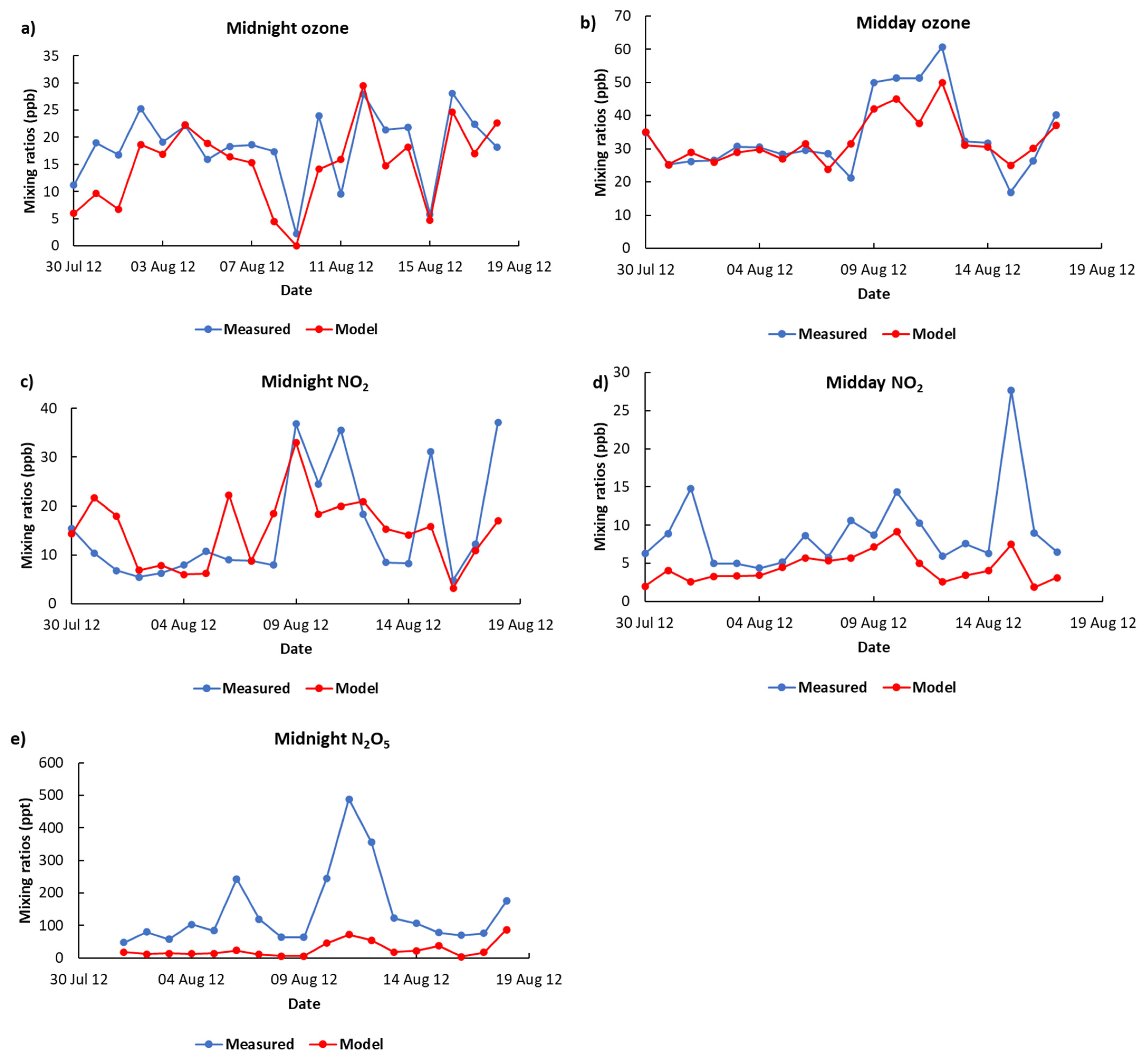
3.1. Model Validation
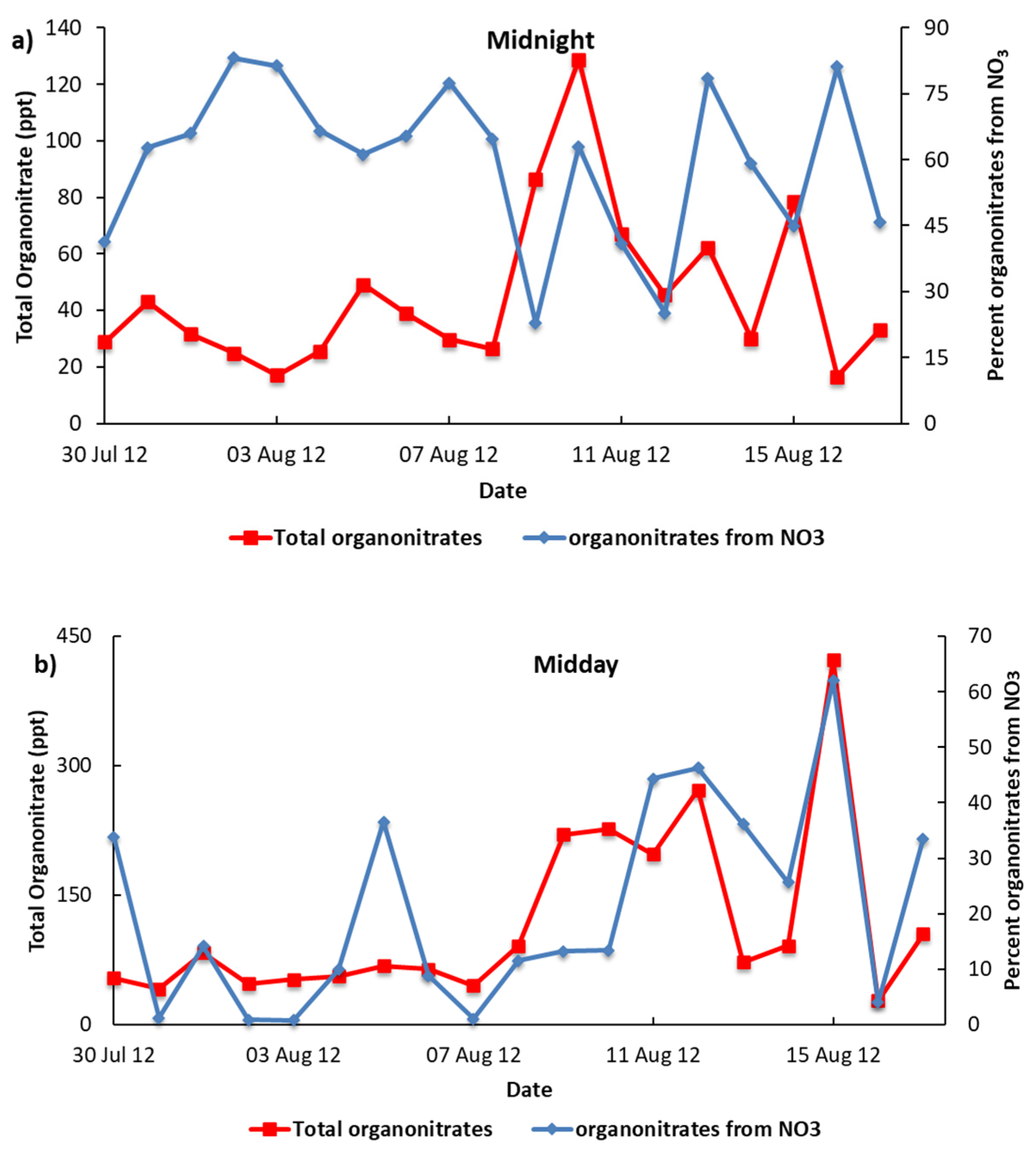
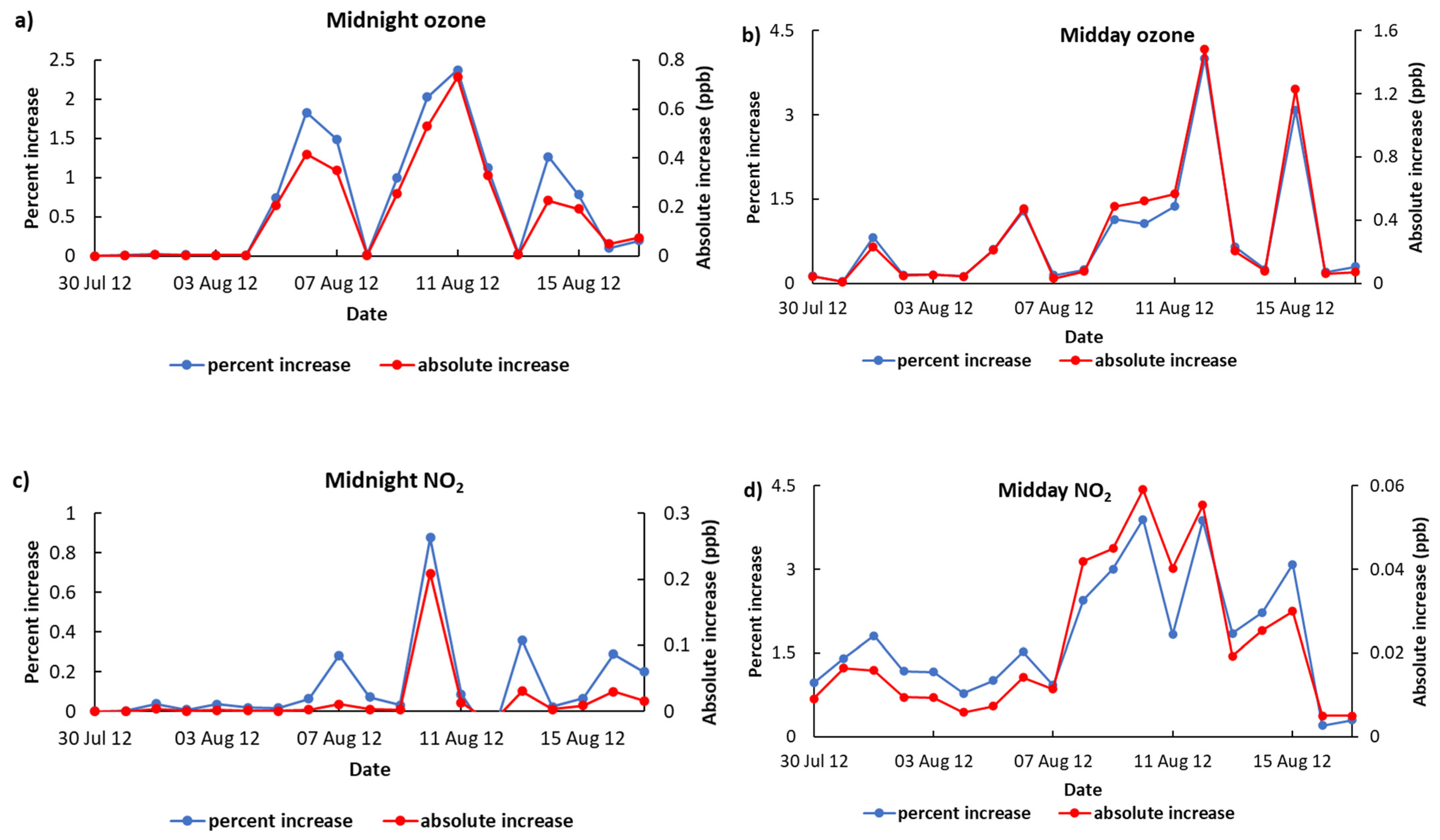
3.2. Contribution of NO3 Sources Organo-Nitrates
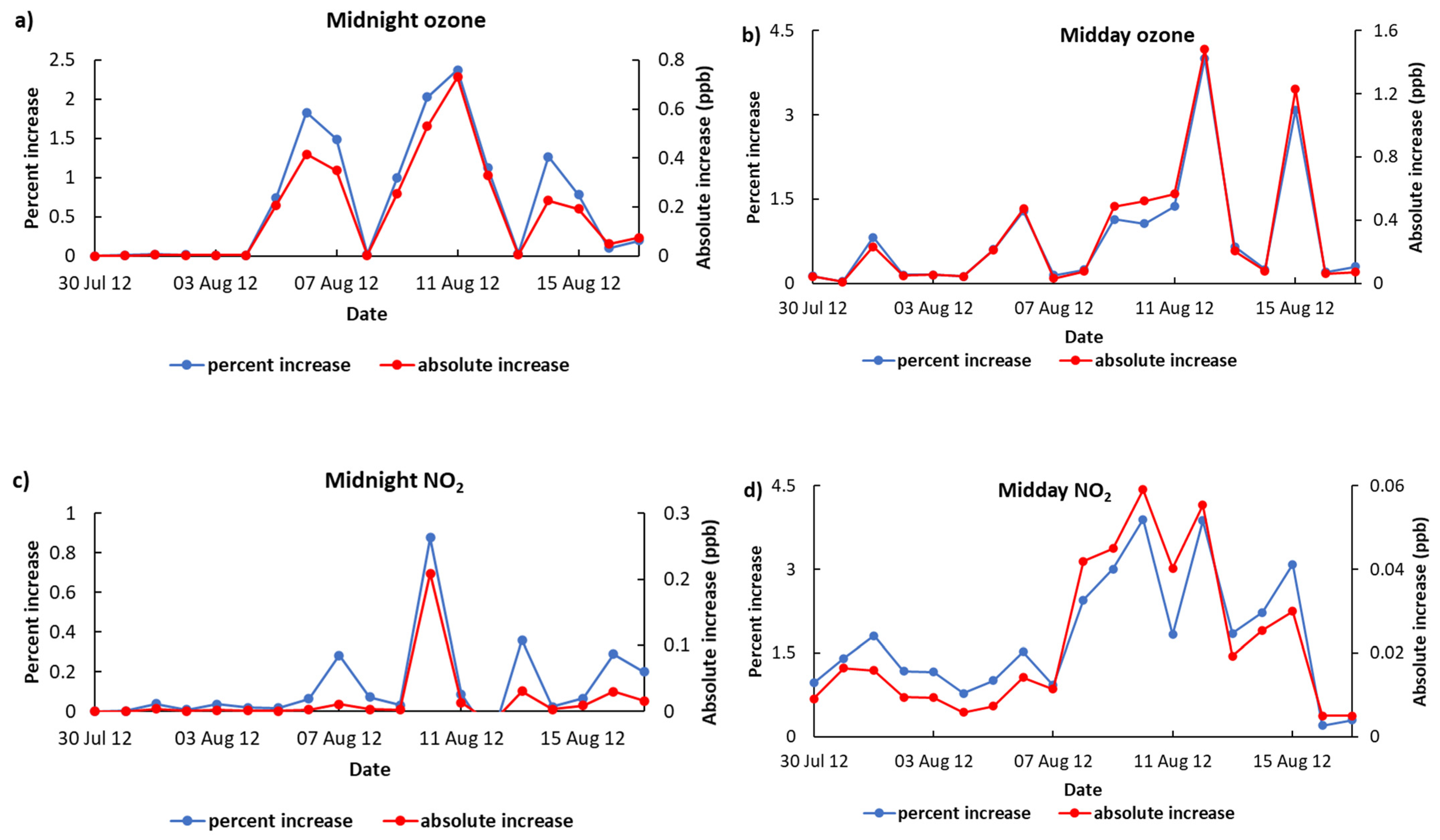
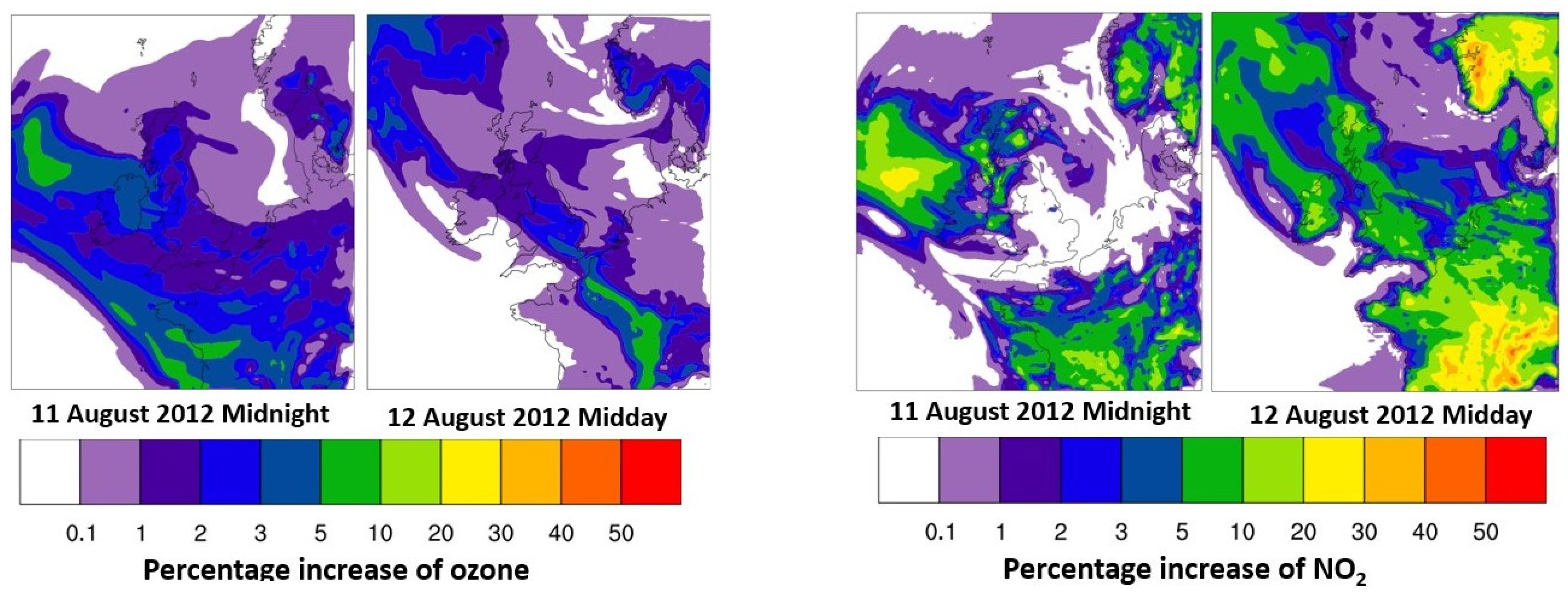
3.3. Atmospheric Implications of NO3-Sourced Organo-Nitrates
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wayne, R.P.; Barnes, I.; Biggs, P.; Burrows, J.P.; Canosas-Mas, C.E.; Hjorth, J.; Le Bras, G.; Moortgat, G.K.; Perner, D.; Poulet, G.; et al. The nitrate radical: Physics, chemistry, and the atmosphere. Atmos. Environ. 1991, 25, 1–203. [Google Scholar] [CrossRef]
- Platt, U.; Alicke, B.; Dubois, R.; Geyer, A.; Hofzumahaus, A.; Holland, F.; Martinez, M.; Mihelcic, D.; Klüpfel, T.; Lohrmann, B.; et al. Free radicals and fast photochemistry during BERLIOZ. J. Atmos. Chem. 2002, 42, 359–394. [Google Scholar] [CrossRef]
- Khan, M.A.H.; Morris, W.C.; Watson, L.A.; Galloway, M.; Hamer, P.D.; Shallcross, B.M.A.; Percival, C.J.; Shallcross, D.E. Estimation of daytime NO3 radical levels in the UK urban atmosphere using the steady state approximation method. Adv. Meteorol. 2015, 2015, 294069. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.H.; Cooke, M.C.; Utembe, S.R.; Archibald, A.T.; Derwent, R.G.; Xiao, P.; Percival, C.J.; Jenkin, M.E.; Morris, W.C.; Shallcross, D.E. Global modelling of the nitrate radical (NO3) for present and pre-industrial scenarios. Atmos. Res. 2015, 164, 347–357. [Google Scholar] [CrossRef]
- Brown, S.S.; Stutz, J. Nighttime radical observations and chemistry. Chem. Soc. Rev. 2012, 41, 6405–6447. [Google Scholar] [CrossRef]
- Brown, S.S.; Ryerson, T.B.; Wollny, A.G.; Brock, C.A.; Peltier, R.; Sullivan, A.P.; Weber, R.J.; Dubé, W.P.; Trainer, M.; Meagher, J.F.; et al. Variability in nocturnal nitrogen oxide processing and its role in regional air quality. Science 2006, 311, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Fish, D.J.; Shallcross, D.E.; Jones, R.L. The vertical distribution of NO3 in the atmospheric boundary layer. Atmos. Environ. 1999, 33, 687–691. [Google Scholar] [CrossRef]
- Stone, D.; Evans, M.J.; Walker, H.; Ingham, T.; Vaughan, S.; Ouyang, B.; Kennedy, O.J.; McLeod, M.W.; Jones, R.L.; Hopkins, J.; et al. Radical chemistry at night: Comparisons between observed and modelled HOx, NO3 and N2O5 during the RONOCO project. Atmos. Chem. Phys. 2014, 14, 1299–1321. [Google Scholar] [CrossRef] [Green Version]
- Aliwell, S.R.; Jones, R.L. Measurements of tropospheric NO3 at midaltitude. J. Geophys. Res. Atmos. 1998, 103, 5719–5727. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications; Academic Press: Cambidge, MA, USA, 2000. [Google Scholar]
- Asaf, D.; Tas, E.; Pedersen, D.; Peleg, M.; Luria, M. Long-Term Measurements of NO3 Radical at a Semiarid Urban Site: 2. Seasonal Trends and Loss Mechanisms. Environ. Sci. Technol. 2010, 44, 5901–5907. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.L.; Brown, S.S.; Archibald, A.T.; Atlas, E.; Cohen, R.C.; Crowley, J.N.; Day, D.A.; Donahue, N.M.; Fry, J.L.; Fuchs, H.; et al. Nitrate radicals and biogenic volatile organic compounds: Oxidation, mechanisms, and organic aerosol. Atmos. Chem. Phys. 2017, 17, 2103–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics; John Wiley and Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Atkinson, R. Kinetics and mechanism of the gas-phase reactions of the NO3 radical with organic compounds. J. Phys. Chem. Ref. Data 1991, 20, 459–507. [Google Scholar] [CrossRef] [Green Version]
- Platt, U.; LeBras, G.; Poulet, G.; Burrows, J.P.; Moortgat, G. Peroxy radicals from night-time reaction of NO3 with organic compounds. Nature 1990, 348, 147–149. [Google Scholar] [CrossRef]
- Geyer, A.; Alicke, B.; Ackermann, R.; Martinez, M.; Harder, H.; BRUNE, W.; di Carlo, P.; Williams, E.; Jobson, T.; Hall, S.; et al. Direct observations of daytime NO3: Implications for urban boundary layer chemistry. J. Geophys. Res. Atmos. 2003, 108, 4368. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Saathoff, H.; Shen, X.; Ramisetty, R.; Leisner, T.; Mohr, C. Chemical Characterization of Highly Functionalized Organonitrates Contribution to Night-Time Organic Aerosol Mass Loadings and Particle Growth. Environ. Sci. Technol. 2019, 53, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Farmer, D.K.; Matsunaga, A.; Docherty, K.S.; Surratt, J.D.; Seinfeld, J.H.; Ziemann, P.J.; Jimenez, J.L. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 6670–6675. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, R.; Aschmann, S.M.; Carter, W.P.; Winer, A.M.; Pitts, J.N., Jr. Alkyl nitrate formation from the nitrogen oxide (NOx)-air photooxidations of C2-C8 n-alkanes. J. Phys. Chem. 1982, 86, 4563–4569. [Google Scholar] [CrossRef]
- Geyer, A.; Ackermann, R.; Dubois, R.; Lohrmann, B.; Müller, R.; Platt, U. Long-term observation of nitrate radicals in the continental boundary layer near Berlin. Atmos. Environ. 2001, 35, 3619–3631. [Google Scholar] [CrossRef]
- Roberts, J.M. The atmospheric chemistry of organic nitrates. Atmos. Environ. 1990, 24, 243–287. [Google Scholar] [CrossRef]
- Clemitshaw, K.C.; Williams, J.; Rattigan, O.V.; Shallcross, D.E.; Law, K.S.; Cox, R.A. Gas-phase ultraviolet absorption cross-sections and atmospheric lifetimes of several C2-C5 alkyl nitrates. J. Photoch. Photobio. A 1997, 102, 117–126. [Google Scholar] [CrossRef]
- Flocke, F.; Volz-Thomas, A.; Buers, H.J.; Patz, W.; Garthe, H.J.; Kley, D. Long-term measurements of alkyl nitrates in southern Germany: 1. General behavior and seasonal and diurnal variation. J. Geophys. Res. 1998, 103, 5729–5746. [Google Scholar] [CrossRef]
- Talukdar, R.K.; Burkholder, J.B.; Hunter, M.; Gilles, M.K.; Roberts, J.M.; Ravishankara, A.R. Atmospheric fate of several alkyl nitrates Part 2 UV absorption cross-sections and photodissociation quantum yields. J. Chem. Soc. Faraday Trans. 1997, 93, 2797–2805. [Google Scholar] [CrossRef]
- Von Kuhlmann, R.; Lawrence, M.G.; Pöschl, U.; Crutzen, P.J. Sensitivities in global scale modeling of isoprene. Atmos. Chem. Phys. 2004, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fiore, A.M.; Horowitz, L.W.; Purves, D.W.; Levy, H.; Evans, M.J.; Wang, Y.; Li, Q.; Yantosca, R.M. Evaluating the contributing of changes in isoprene emissions to surface ozone trends over the eastern United States. J. Geophys. Res. Atmos. 2005, 110, D12303. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, L.W.; Fiore, A.M.; Milly, G.P.; Cohen, R.C.; Perring, A.; Wooldridge, P.J.; Hess, P.G.; Emmons, L.K.; Lamarque, J.-F. Observational constraints on the chemistry of isoprene nitrates over the eastern United States. J. Geophys. Res. Atmos. 2007, 112, D12S08. [Google Scholar] [CrossRef]
- Perring, A.E.; Bertram, T.H.; Farmer, D.K.; Wooldridge, P.J.; Dibb, J.; Blake, N.J.; Blake, D.R.; Singh, H.B.; Fuelberg, H.; Diskin, G.; et al. The production and persistence of ΣRONO2 in the Mexico City plume. Atmos. Chem. Phys. 2010, 10, 7215–7229. [Google Scholar] [CrossRef] [Green Version]
- Farmer, D.K.; Perring, A.E.; Wooldridge, P.J.; Blake, D.R.; Baker, A.; Meinardi, S.; Huey, L.G.; Tanner, D.; Vargas, O.; Cohen, R.C. Impact of organic nitrates on urban ozone production. Atmos. Chem. Phys. 2011, 11, 4085–4094. [Google Scholar] [CrossRef] [Green Version]
- Paulot, F.; Henze, D.K.; Wennberg, P.O. Impact of the isoprene photochemical cascade on tropical ozone. Atmos. Chem. Phys. 2012, 12, 1307–1325. [Google Scholar] [CrossRef] [Green Version]
- Griffin, R.J.; Cocker, D.R.; Flagan, R.C.; Seinfeld, J.H. Organic aerosol formation from the oxidation of biogenic hydrocarbons. J. Geophys. Res. Atmos. 1999, 104, 3555–3567. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Goldstein, A.H.; Kroll, J.H.; Ng, N.L.; Varutbangkul, V.; Flagan, R.C.; Seinfeld, J.H. Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes. J. Geophys. Res. Atmos. 2006, 111, D17305. [Google Scholar] [CrossRef] [Green Version]
- Rollins, A.W.; Kiendler-Scharr, A.; Fry, J.L.; Brauers, T.; Brown, S.S.; Dorn, H.-P.; Dubé, W.P.; Fuchs, H.; Mensah, A.; Mentel, T.F.; et al. Isoprene oxidation by nitrate radical: Alkyl nitrate and secondary organic aerosol yields. Atmos. Chem. Phys. 2009, 9, 6685–6703. [Google Scholar] [CrossRef] [Green Version]
- Slade, J.H.; de Perre, C.; Lee, L.; Shepson, P.B. Nitrate radical oxidation of γ -terpinene: Hydroxy nitrate, total organic nitrate, and secondary organic aerosol yields. Atmos. Chem. Phys. 2017, 17, 8635–8650. [Google Scholar] [CrossRef] [Green Version]
- Fry, J.L.; Brown, S.S.; Middlebrook, A.M.; Edwards, P.M.; Campuzano-Jost, P.; Day, D.A.; Jimenez, J.L.; Allen, H.M.; Ryerson, T.B.; Pollack, I.; et al. Secondary organic aerosol (SOA) yields from NO3 radical + isoprene based on nighttime aircraft power plant plume transects. Atmos. Chem. Phys. 2018, 18, 11663–11682. [Google Scholar] [CrossRef] [Green Version]
- Ng, N.L.; Kwan, A.J.; Surratt, J.D.; Chan, A.W.H.; Chhabra, P.S.; Sorooshian, A.; Pye, H.O.T.; Crounse, J.D.; Wennberg, P.O.; Flagan, R.C.; et al. Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3). Atmos. Chem. Phys. 2008, 8, 4117–4140. [Google Scholar] [CrossRef] [Green Version]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef] [Green Version]
- IPCC. Climate Change 2013: The Physical Scientific Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; p. 1535. [Google Scholar]
- Nel, A. Air pollution-related illness: Effects of particles. Science 2005, 308, 804–806. [Google Scholar] [CrossRef]
- Sharaiwa, M.; Ueda, K.; Pozzer, A.; Lammel, G.; Kampf, C.J.; Fushimi, A.; Enami, S.; Arangio, A.M.; Fröhlich-Nowoisky, J.; Fujitani, Y.; et al. Aerosol Heatlth Effects from Molecular to Global Scales. Environ. Sci. Technol. 2017, 51, 13545–13567. [Google Scholar] [CrossRef]
- Rollins, A.W.; Browne, E.C.; Min, K.-E.; Pusede, S.E.; Wooldridge, P.J.; Gentner, D.R.; Goldstein, A.H.; Liu, S.; Day, D.A.; Russell, L.M.; et al. Evidence for NOx control over nighttime SOA formation. Science 2012, 337, 1210–1212. [Google Scholar] [CrossRef]
- Kiendler-Scharr, A.; Mensah, A.A.; Friese, E.; Topping, D.; Nemitz, E.; Prevot, A.S.H.; Äijälä, M.; Allan, J.; Canonaco, F.; Canagaratna, M.; et al. Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol. Geophys. Res. Lett. 2016, 43, 7735–7744. [Google Scholar] [CrossRef] [Green Version]
- Bohnenstengel, S.I.; Belcher, S.E.; Aiken, A.; Allan, J.D.; Allen, G.; Bacak, A.; Bannan, T.J.; Barlow, J.F.; Beddows, D.C.S.; Bloss, W.J.; et al. Meteorology, Air Quality, and Health in London: The ClearfLo Project. Bull. Am. Meteorol. Soc. 2015, 96, 779–804. [Google Scholar] [CrossRef] [Green Version]
- Bannan, T.J.; Bacak, A.; Muller, J.B.; Booth, A.M.; Jones, B.; Le Breton, M.; Leather, K.E.; Ghalaieny, M.; Xiao, P.; Shallcross, D.E.; et al. Importance of direct anthropogenic emissions of formic acid measured by a chemical ionisation mass spectrometer (CIMS) during the Winter ClearfLo Campaign in London, January 2012. Atmos. Environ. 2014, 83, 301–310. [Google Scholar] [CrossRef]
- Bannan, T.J.; Booth, A.M.; Bacak, A.; Muller, J.B.A.; Leather, K.E.; Le Breton, M.; Jones, B.; Young, D.; Coe, H.; Allan, J.; et al. The first UK measurements of nitryl chloride using a chemical ionisation mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation. J. Geophys. Res. Atmos. 2015, 120, 5638–5657. [Google Scholar] [CrossRef]
- Bannan, T.J.; Bacak, A.; Le Breton, M.; Flynn, M.; Ouyang, B.; McLeod, M.; Jones, R.; Malkin, T.L.; Whalley, L.K.; Heard, D.E.; et al. Ground and airborne UK measurements of nitryl chloride: An investigation of the role of Cl atom oxidation at Weybourne Atmospheric Observatory. J. Geophys. Res. Atmos. 2017, 122, 11–154. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, T.; Yan, C.; Tham, Y.J.; Xue, L.; Xu, Z.; Zha, Q. Large daytime signals of N2O5 and NO3 inferred at 62 amu in a TD-CIMS: Chemical interference or a real atmospheric phenomenon? Atmos. Meas. Tech. 2014, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Le Breton, M.; Bacak, A.; Muller, J.B.A.; Bannan, T.J.; Kennedy, O.; Ouyang, B.; Xiao, P.; Ashfold, M.N.R.; Bauguitte, S.J.-B.; Shallcross, D.E.; et al. The first airborne inter-comparison of N2O5 measurements over the UK using a Chemical Ionisation Mass Spectrometer (CIMS) and Broadband Cavity Enhanced Absorption Spectrometer (BBCEAS) during the RONOCO 2010/2011 campaign. Anal. Meth. 2014, 6, 9731–9743. [Google Scholar] [CrossRef] [Green Version]
- Grell, G.A.; Peckham, S.E.; Schmitz, R.; McKeen, S.A.; Frost, G.; Skamarock, W.C.; Eder, B. Fully coupled “online” chemistry within the WRF model. Atmos. Environ. 2005, 39, 6957–6975. [Google Scholar] [CrossRef]
- Dee, D.P.; Uppala, S.M.; Simmons, A.J.; Berrisford, P.; Poli, P.; Kobayashi, S.; Andrae, U.; Balmaseda, M.A.; Balsamo, G.; Bauer, P.; et al. The ERA-interim reanalysis: Configuration and performance of the data assimilation system. Q. J. R. Meteorol. Soc. 2011, 137, 553–597. [Google Scholar] [CrossRef]
- Emmons, L.K.; Walters, S.; Hess, P.G.; Lamarque, J.-F.; Pfizer, G.G.; Fillmore, D.; Granier, C.; Guenther, A.; Kinnison, D.; Laepple, T.; et al. Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART-4). Geosci. Model. Dev. 2010, 3, 43–67. [Google Scholar] [CrossRef] [Green Version]
- Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, P.; Palmer, P.I.; Geron, C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar] [CrossRef] [Green Version]
- Sakulyanontvittaya, T.; Duhl, T.; Wiedinmyer, C.; Helmig, D.; Matsunaga, S.; Potosnak, M.; Milford, J.; Guenther, A. Monoterpene and sesquiterpene emission estimates for the United States. Environ. Sci. Technol. 2008, 42, 1623–1629. [Google Scholar] [CrossRef] [Green Version]
- Kuenen, J.J.P.; Visschedijk, A.J.H.; Jozwicka, M.; Denier van der Gon, H.A.C. TNO-MACC_IIemission inventory; a multi-year (2003–2009) consistent high-resolution European emission inventory for air quality modelling. Atmos. Chem. Phys. 2014, 14, 10963–10976. [Google Scholar] [CrossRef] [Green Version]
- Jenkin, M.E.; Watson, L.A.; Utembe, S.R.; Shallcross, D.E. A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 1: Gas phase mechanism development. Atmos. Environ. 2008, 42, 7185–7195. [Google Scholar] [CrossRef]
- Watson, L.A.; Shallcross, D.E.; Utembe, S.R.; Jenkin, M.E. A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 2: Gas phase mechanism reduction. Atmos. Environ. 2008, 42, 7196–7204. [Google Scholar] [CrossRef]
- Wild, O.; Zhu, X.; Prather, M.J. Fast-J: Accurate simulation of IN- and Below-Cloud Photolysis in Tropospheric Chemical Models. J. Atmos. Chem. 2000, 37, 245–282. [Google Scholar] [CrossRef]
- Khan, M.A.H.; Clements, J.; Lowe, D.; McFiggans, G.; Percival, C.J.; Shallcross, D.E. Investigating the behaviour of the CRI-MECH gas-phase chemistry scheme on a regional scale for different seasons using the WRF-Chem model. Atmos. Res. 2019, 229, 145–156. [Google Scholar] [CrossRef]
- Archer-Nicholls, S.; Lowe, D.; Utembe, S.; Allan, J.; Zaveri, R.A.; Fast, J.D.; Hodnebrog, Ø.; van der Gon, H.D.; McFiggans, G. Gaseous chemistry and aerosol mechanism developments for version 3.5.1 of the online regional model, WRF-Chem. Geosci. Model. Dev. 2014, 7, 2557–2579. [Google Scholar] [CrossRef] [Green Version]
- Dörich, R.; Eger, P.; Lelieveld, J.; Crowley, J.N. Iodide CIMS and m/z 62: The detection of HNO3 as NO3- in the presence of PAN, peroxyacetic acid and ozone. Atmos. Meas. Tech. 2021, 14, 5319–5332. [Google Scholar] [CrossRef]
- Liebmann, J.; Karu, E.; Sobanski, N.; Schuladen, J.; Ehn, M.; Schallhart, S.; Quéléver, L.; Hellen, H.; Hakola, H.; Hoffmann, T.; et al. Direct measurement of NO3 radical reactivity in a boreal forest. Atmos. Chem. Phys. 2018, 18, 3799–3815. [Google Scholar] [CrossRef] [Green Version]
- Liebmann, J.M.; Muller, J.B.A.; Kubistin, D.; Claude, A.; Holla, R.; Plass-Dülmer, C.; Lelieveld, J.; Crowley, J.N. Direct measurements of NO3 reactivity in and above the boundary layer of a mountaintop site: Identification of reactive trace gases and comparison with OH reactivity. Atmos. Chem. Phys. 2018, 18, 12045–12059. [Google Scholar] [CrossRef] [Green Version]
- Kames, J.; Schurath, U. Alkyl nitrates and bifunctional nitrates of atmospheric interest: Henry’s law constants and their temperature dependencies. J. Atmos. Chem. 1992, 15, 79–95. [Google Scholar] [CrossRef]
- Utembe, S.R.; Cooke, M.C.; Archibald, A.T.; Shallcross, D.E.; Derwent, R.G.; Jenkin, M.E. Simulating secondary organic aerosol in a 3-D Lagrangian chemistry transport model using the reduced Common Representative Intermediates mechanism (CRI v2-R5). Atmos. Environ. 2011, 45, 1604–1614. [Google Scholar] [CrossRef]
- Khan, M.A.H.; Jenkin, M.E.; Foulds, A.; Derwent, R.G.; Percival, C.J.; Shallcross, D.E. A modeling study of secondary organic aerosol formation from sesquiterpenes using the STOCHEM global chemistry and transport model. J. Geophys. Res. Atmos. 2017, 122, 4426–4439. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organo-Nitrate Species * | Production Pathway |
|---|---|
| CH3NO3 (CH3NO3) | CH3O2 + NO |
| C2H5NO3 (C2H5NO3) | C2H5O2 + NO |
| IC3H7NO3 (IC3H7NO3) | IC3H7O2 + NO |
| HOC2H4NO3 (HOC2H4NO3) | HOCH2CH2O2+ NO |
| NRU12O2 (C510O2, NC4CO3) | C5H8 + NO3 |
| NRU12OOH (C510OOH, NC4CO3H) | C5H8 + NO3 |
| NRN6O2 (ETHENO3O2) | C2H4 + NO3 |
| NRN6OOH (ETHO2HNO3) | C2H4 + NO3 |
| NRN9O2 (PRONO3AO2, PRONO3BO2) | C3H6 + NO3 |
| NRN9OOH (PR1O2HNO3, PR2O2HNO3) | C3H6 + NO3 |
| NRN12O2 (C42NO33O2) | TBUT2ENE + NO3 |
| NRN12OOH (C42NO33OOH) | TBUT2ENE + NO3 |
| NOA (NOA) | C5H8 + NO3 |
| RN10NO3 (NC3H7NO3) | RN10O2 (NC3H7O2) + NO |
| RN13NO3 (NC4H9NO3, SC4H9NO3) | RN13O2 (NC4H9O2, SC4H9O2) + NO |
| RN19NO3 (HEXCNO3, M2PEDNO3, M3PECNO3) | RN19O2 (HEXCO2, M2PEDO2, M3PECO2) + NO |
| RN9NO3 (PROPOLNO3, PROLNO3) | RN9O2 (HYPROPO2, IPROPOLO2) + NO |
| RN12NO3 (HO1C4NO3, BUT2OLNO3) | RN12O2 (HO1C4O2, BUT2OLO2) + NO |
| RN15NO3 (PE1ENEANO3, PE2ENEANO3, HO2C5NO3) | RN15O2 (PE1ENEAO2, PE2ENEAO2, HO2C5O2) + NO |
| RN18NO3 (C65OH4NO3, C6OH5NO3, HO2C6NO3) | RN18O2 (C65OH4O2, C6OH5O2, HO2C6O2) + NO |
| RN16NO3 (PEANO3, PEBNO3, PECNO3) | RN16O2 (PEAO2, PEBO2, PECO2) + NO |
| RU14NO3 (ISOPANO3, ISOPBNO3, ISOPCNO3, ISOPDNO3) | RU14O2 (ISOPAO2, ISOPBO2, ISOPCO2, ISOPDO2) + NO |
| RA13NO3 (BZBIPERNO3) | RA13O2 (BZBIPERO2) + NO |
| RA16NO3 (TLBIPERNO3) | RA16O2 (TLBIPERO2) + NO |
| RA19NO3 (OXYBIPENO3) | RA19AO2 (OXYBIPERO2) + NO |
| RA25NO3 (DM35EBNO3) | RA25O2 (DM35EBO2) + NO |
| RA22NO3 (TM123BNO3) | RA22AO2 (TM123BO2) + NO |
| RTN28NO3 (APINANO3, APINBNO3, APINCNO3) | RTN28O2 (APINAO2, APINBO2, APINCO2) + NO |
| NRTN28O2 (NAPINAO2, NAPINBO2) | APINENE + NO3 |
| NRTN28OOH (NAPINAOOH, NAPINBOOH) | APINENE + NO3 |
| RTN25NO3 (C96NO3) | RTN25O2 (C96O2) + NO |
| RTN23NO3 (C98NO3) | RTN23O2 (C98O2) + NO |
| RTX24NO3 (NOPINANO3, NOPINBNO3, NOPINCNO3) | RTX24O2 (NOPINAO2, NOPINBO2, NOPINCO2) + NO |
| RTX22NO3 (C915NO3, C917NO3, C918NO3) | RTX22O2 (C915O2, C917O2, C918O2) + NO |
| RTX28NO3 (BPINANO3, BPINBNO3, BPINCNO3) | RTX28O2 (BPINAO2, BPINBO2, BPINCO2) + NO |
| NRTX28O2 (NBPINAO2, NBPINBO2) | BPINENE + NO3 |
| NRTX28OOH (NBPINAOOH, NBPINBOOH) | BPINENE + NO3 |
| NRU14O2 (NISOPO2) | C5H8 + NO3 |
| NRU14OOH (NISOPOOH) | C5H8 + NO3 |
| Organo-Nitrates | Lifetime (July–August) (Days) | |
|---|---|---|
| OH | Photolysis | |
| NRN12OOH | 1.0 | 10.7 |
| NRN9OOH | 1.3 | 11.1 |
| NRN6OOH | 1.7 | 10.9 |
| NRU12OOH | 0.3 | 5.1 |
| NRU14OOH | 0.4 | 36.5 |
| NRTN28OOH | 0.8 | 9.0 |
| NRTX28OOH | 0.7 | 7.4 |
| NOA | 42.1 | 17.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foulds, A.; Khan, M.A.H.; Bannan, T.J.; Percival, C.J.; Lowenberg, M.H.; Shallcross, D.E. Abundance of NO3 Derived Organo-Nitrates and Their Importance in the Atmosphere. Atmosphere 2021, 12, 1381. https://doi.org/10.3390/atmos12111381
Foulds A, Khan MAH, Bannan TJ, Percival CJ, Lowenberg MH, Shallcross DE. Abundance of NO3 Derived Organo-Nitrates and Their Importance in the Atmosphere. Atmosphere. 2021; 12(11):1381. https://doi.org/10.3390/atmos12111381
Chicago/Turabian StyleFoulds, Amy, M. Anwar H. Khan, Thomas J. Bannan, Carl J. Percival, Mark H. Lowenberg, and Dudley E. Shallcross. 2021. "Abundance of NO3 Derived Organo-Nitrates and Their Importance in the Atmosphere" Atmosphere 12, no. 11: 1381. https://doi.org/10.3390/atmos12111381
APA StyleFoulds, A., Khan, M. A. H., Bannan, T. J., Percival, C. J., Lowenberg, M. H., & Shallcross, D. E. (2021). Abundance of NO3 Derived Organo-Nitrates and Their Importance in the Atmosphere. Atmosphere, 12(11), 1381. https://doi.org/10.3390/atmos12111381