Effect of Renewable Fuels and Intake O2 Concentration on Diesel Engine Emission Characteristics and Reactive Oxygen Species (ROS) Formation

 , , , , and
, , , , and
Abstract
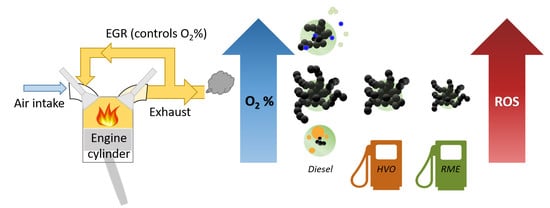
1. Introduction
2. Materials and Methods
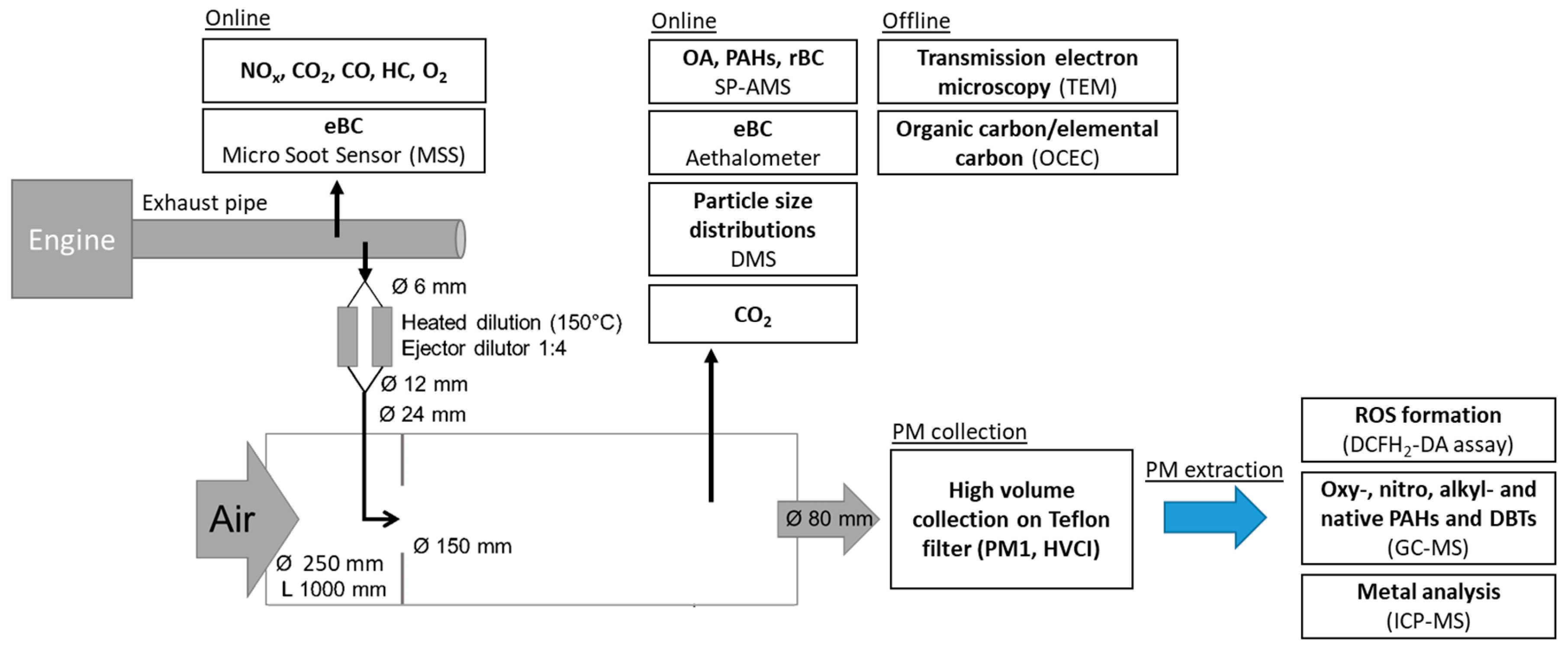
2.1. Emission Generation
2.2. Dilution System
2.3. Emission Characterization
2.3.1. Thermal-Optical Carbon Analysis and Transmission Electron Microscopy Imaging
2.3.2. SP-AMS
2.4. PM Collection with HVCI and Extraction for ROS, PAH and Metal Analysis
2.4.1. ROS Assay
2.4.2. PAH Analysis
2.4.3. Metal Analysis
3. Results and Discussion
3.1. Exhaust Emissions
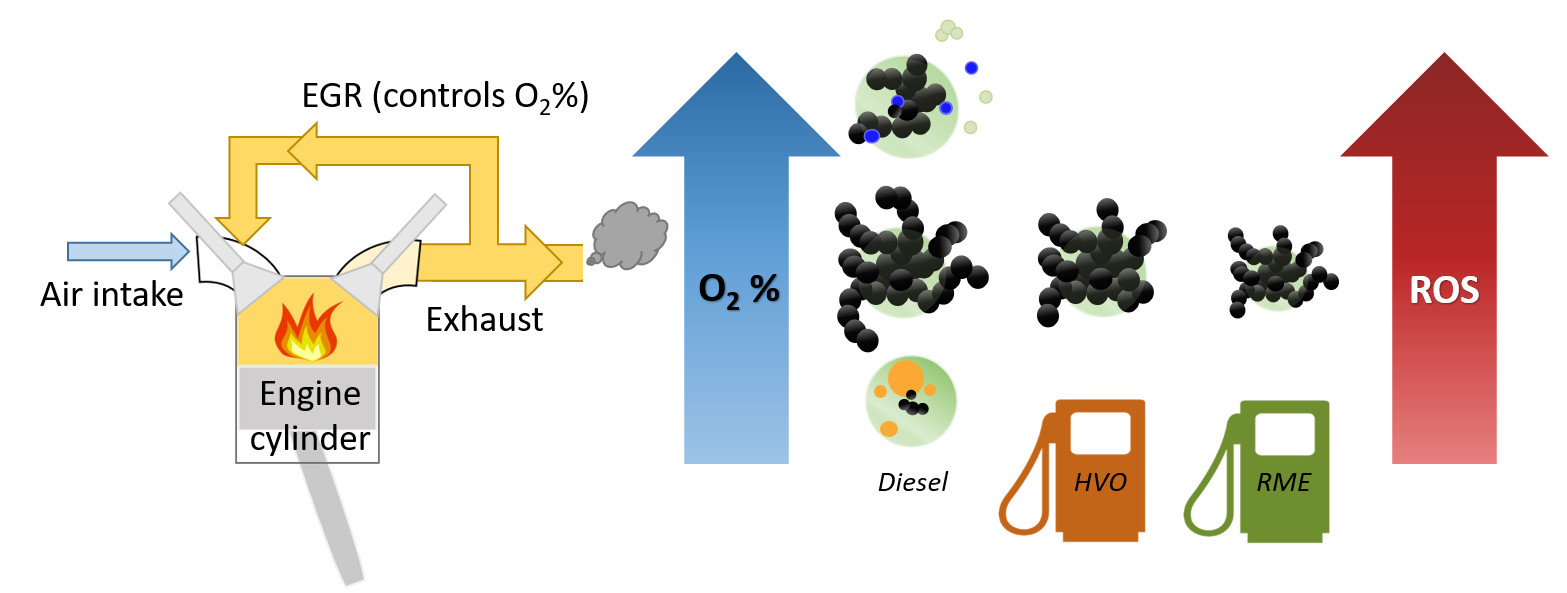
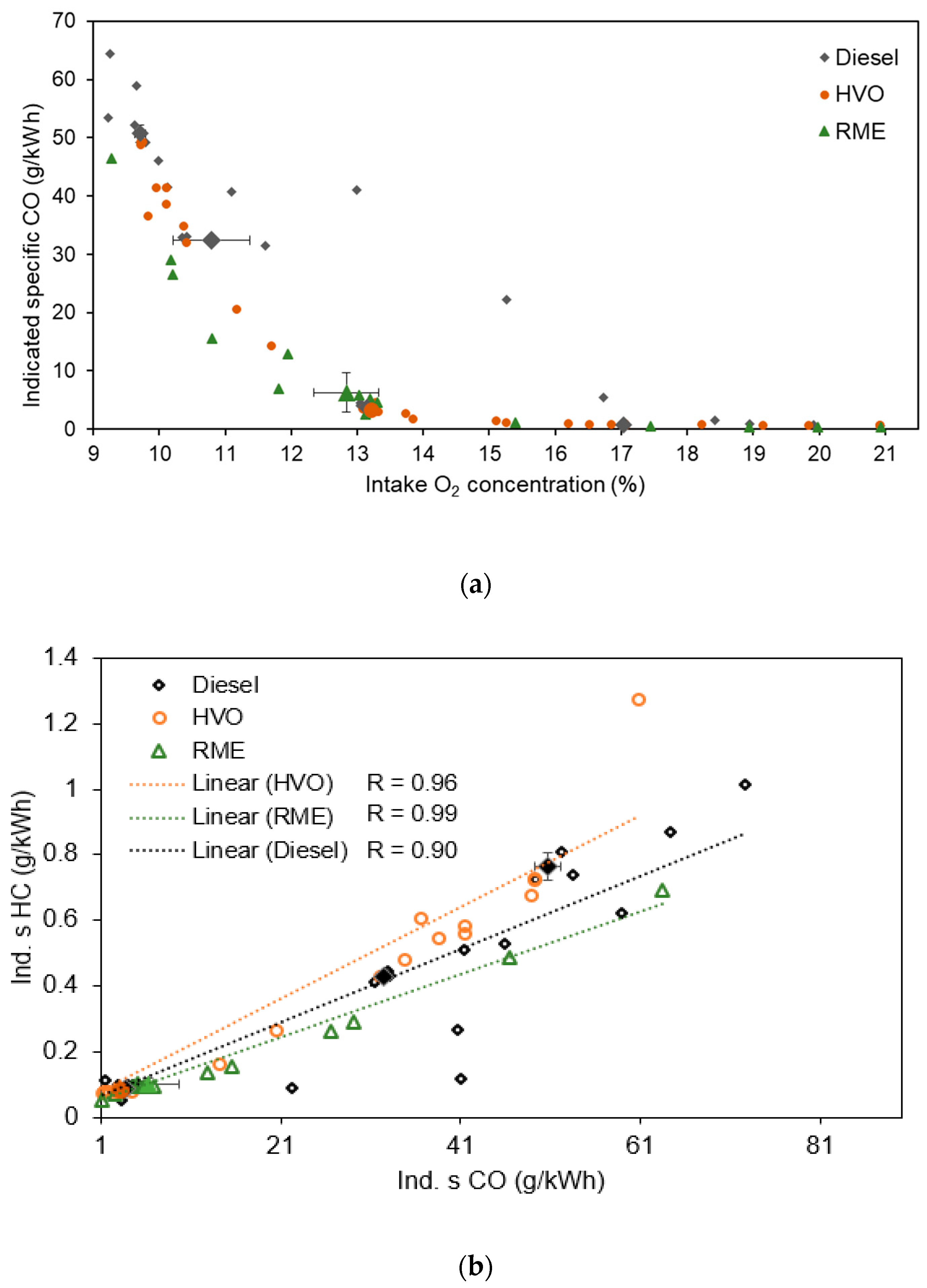
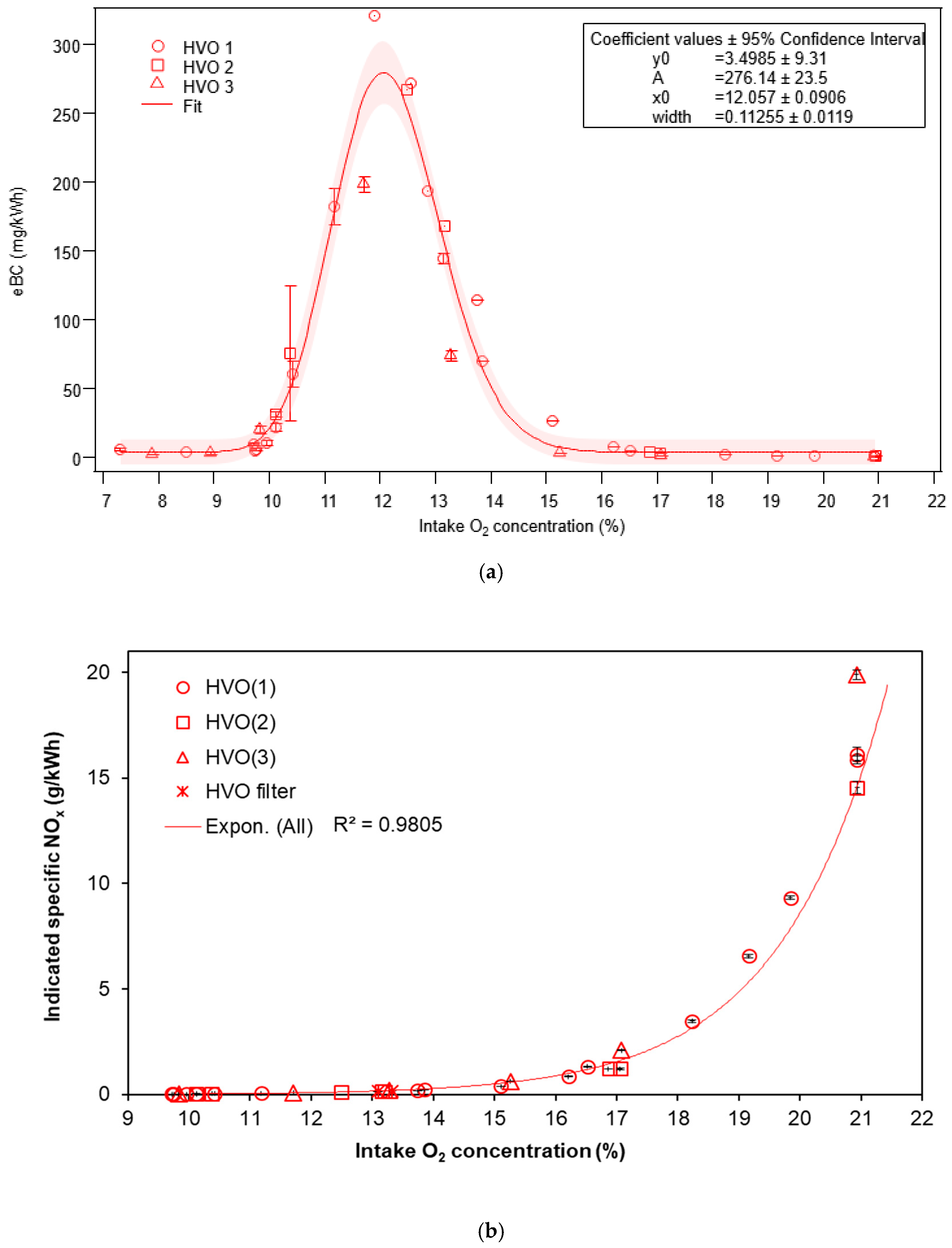
3.1.1. Equivalent Black Carbon and Regulated Emissions
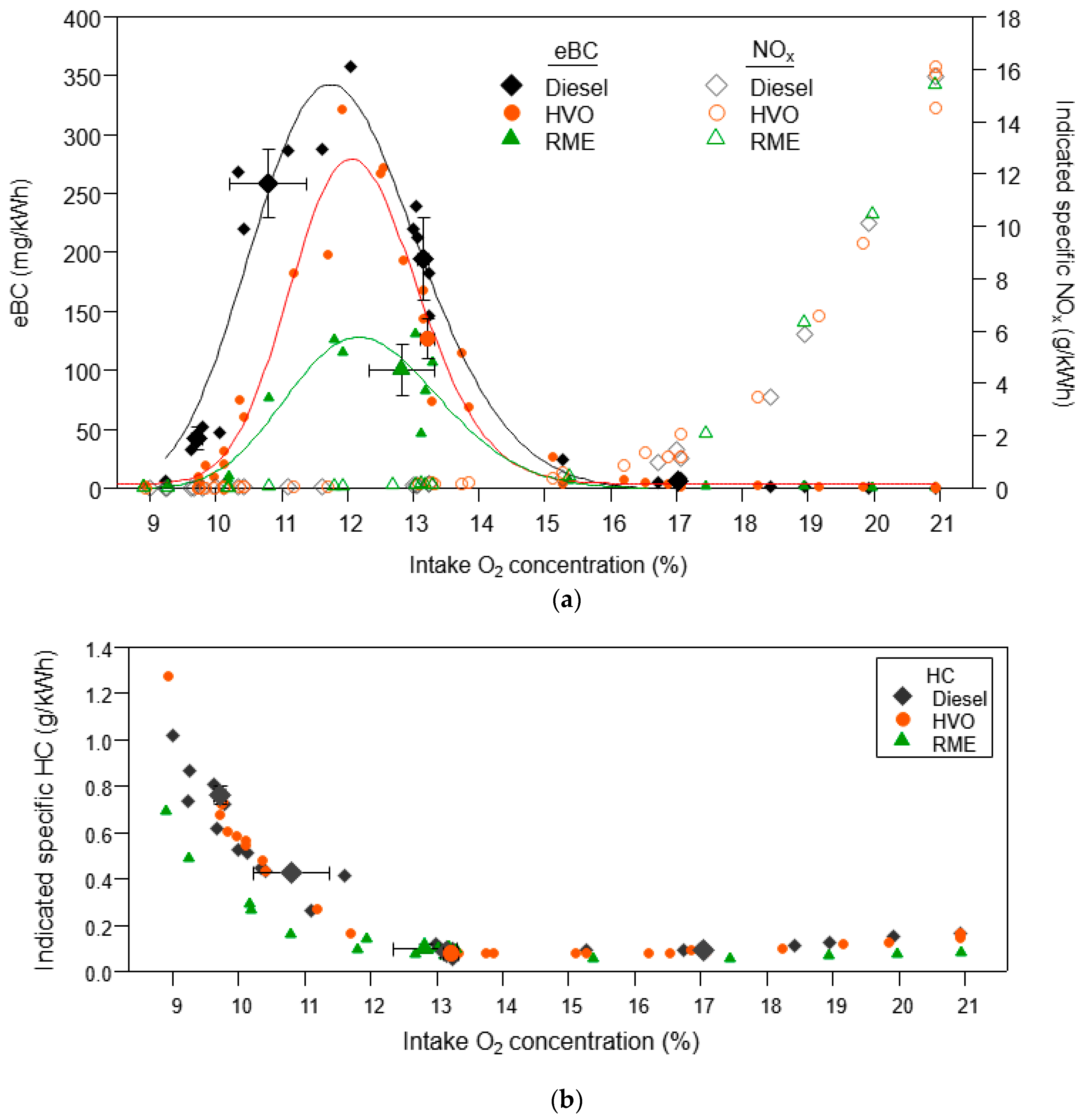
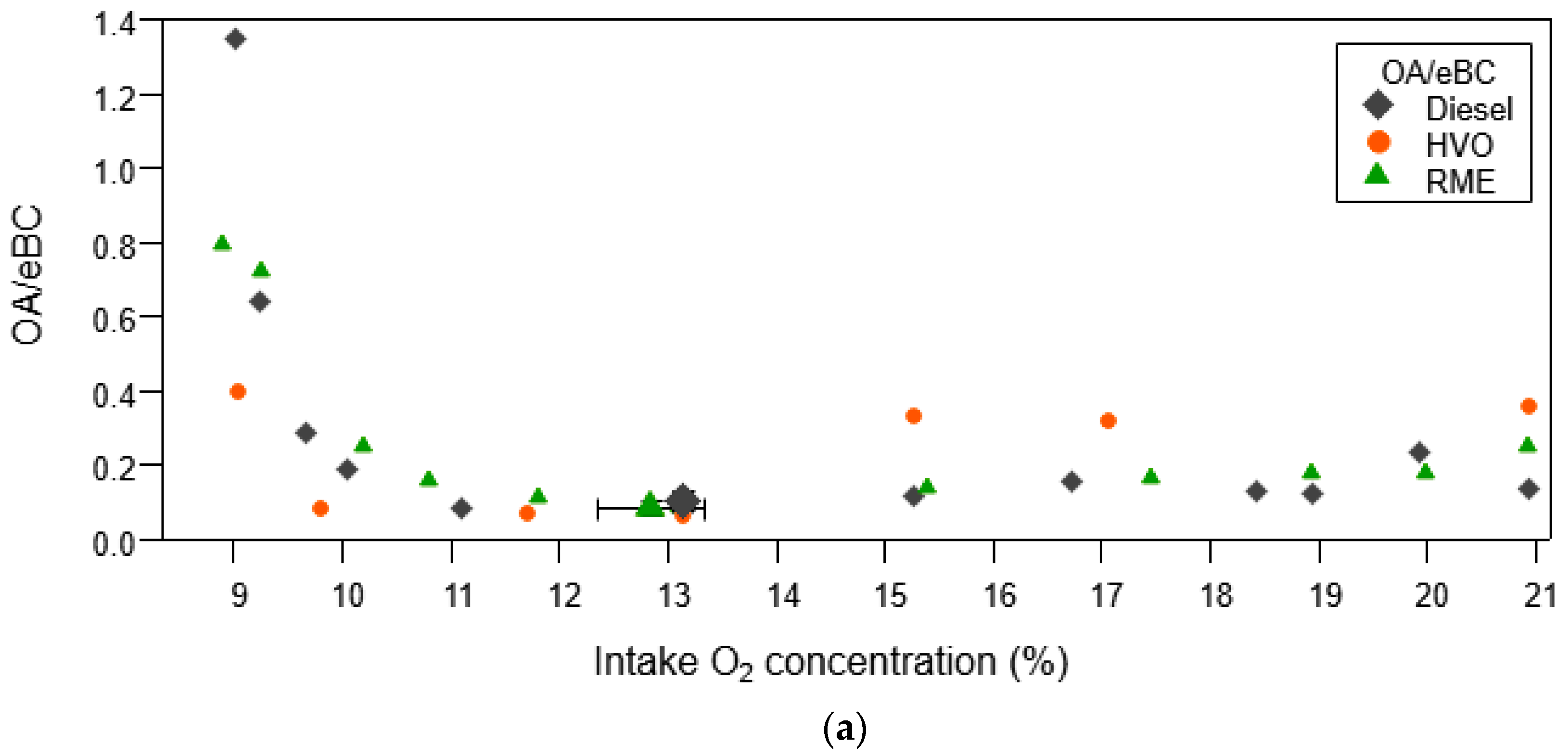
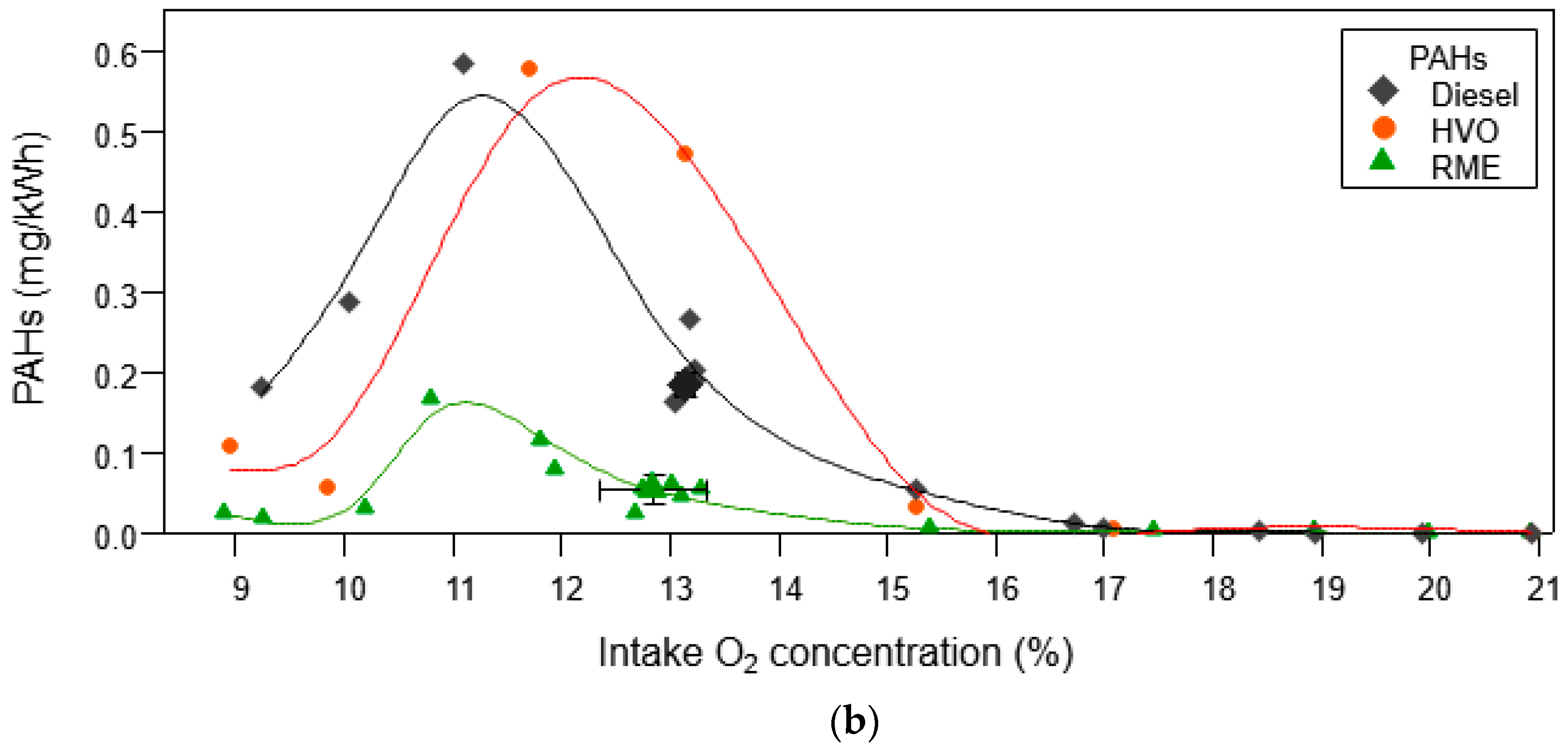
3.1.2. Organic Aerosol and PAH Emissions
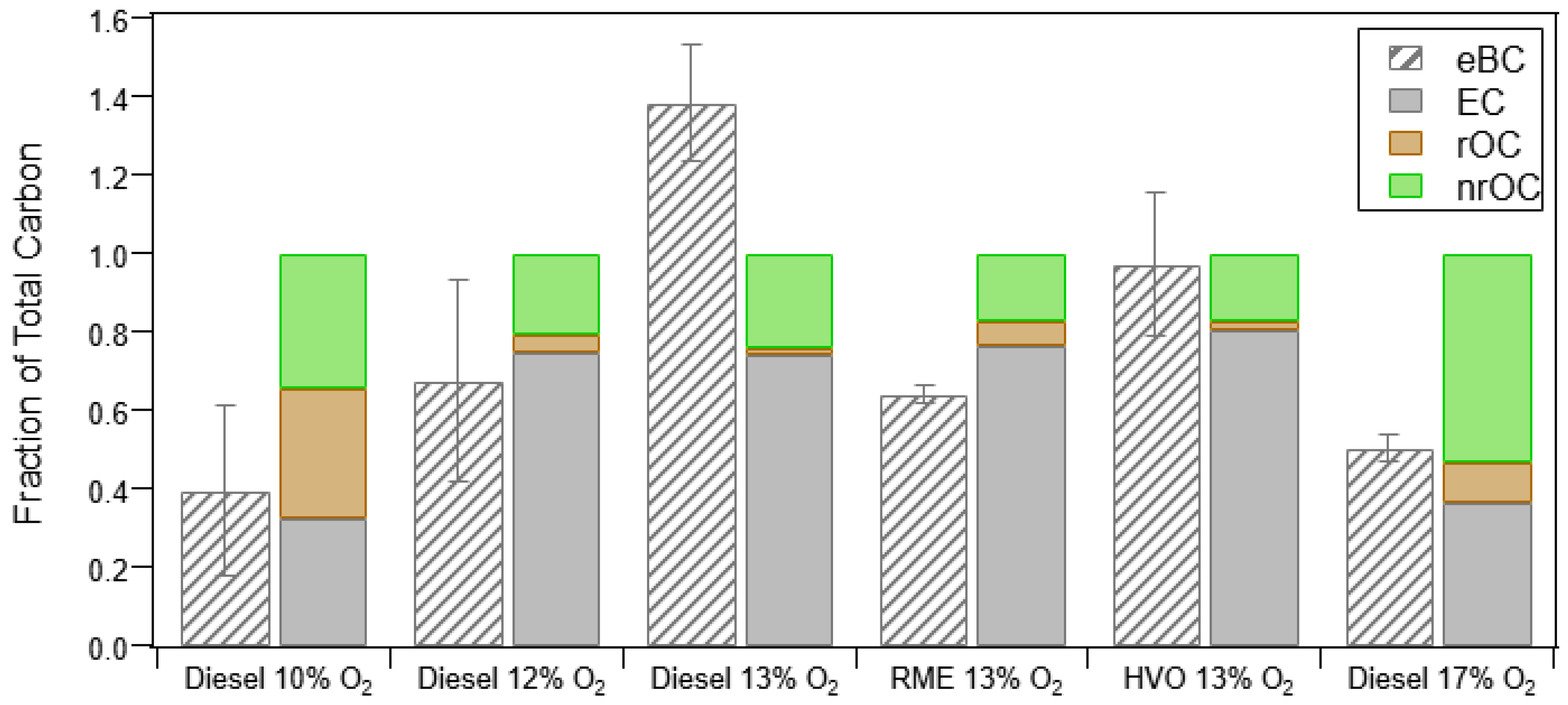
3.1.3. Thermal Optical Analysis (OC/EC) and Relationship to PM Measurements
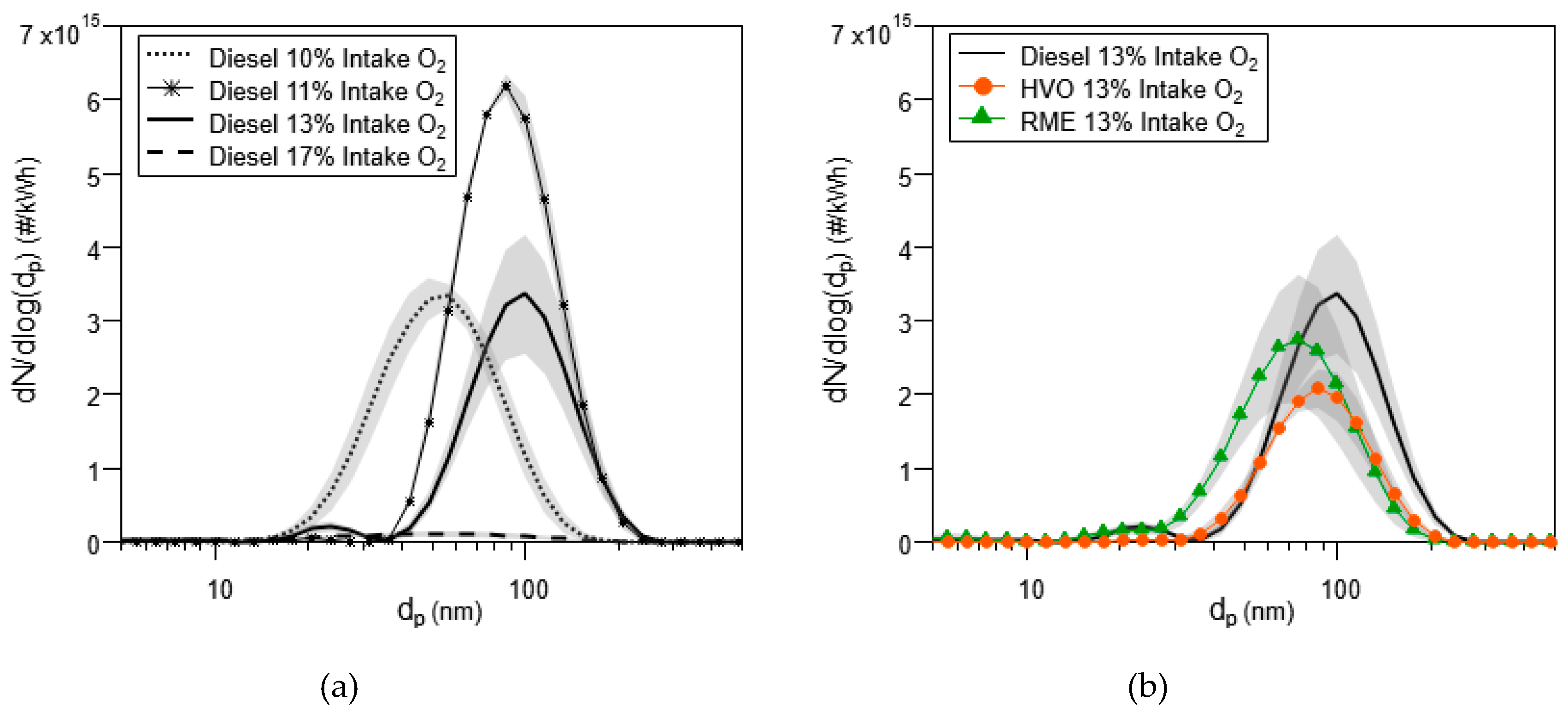
3.1.4. Particle Size Distributions and TEM Analysis
3.2. Analysis of Extracted PM for PAHs and Metals
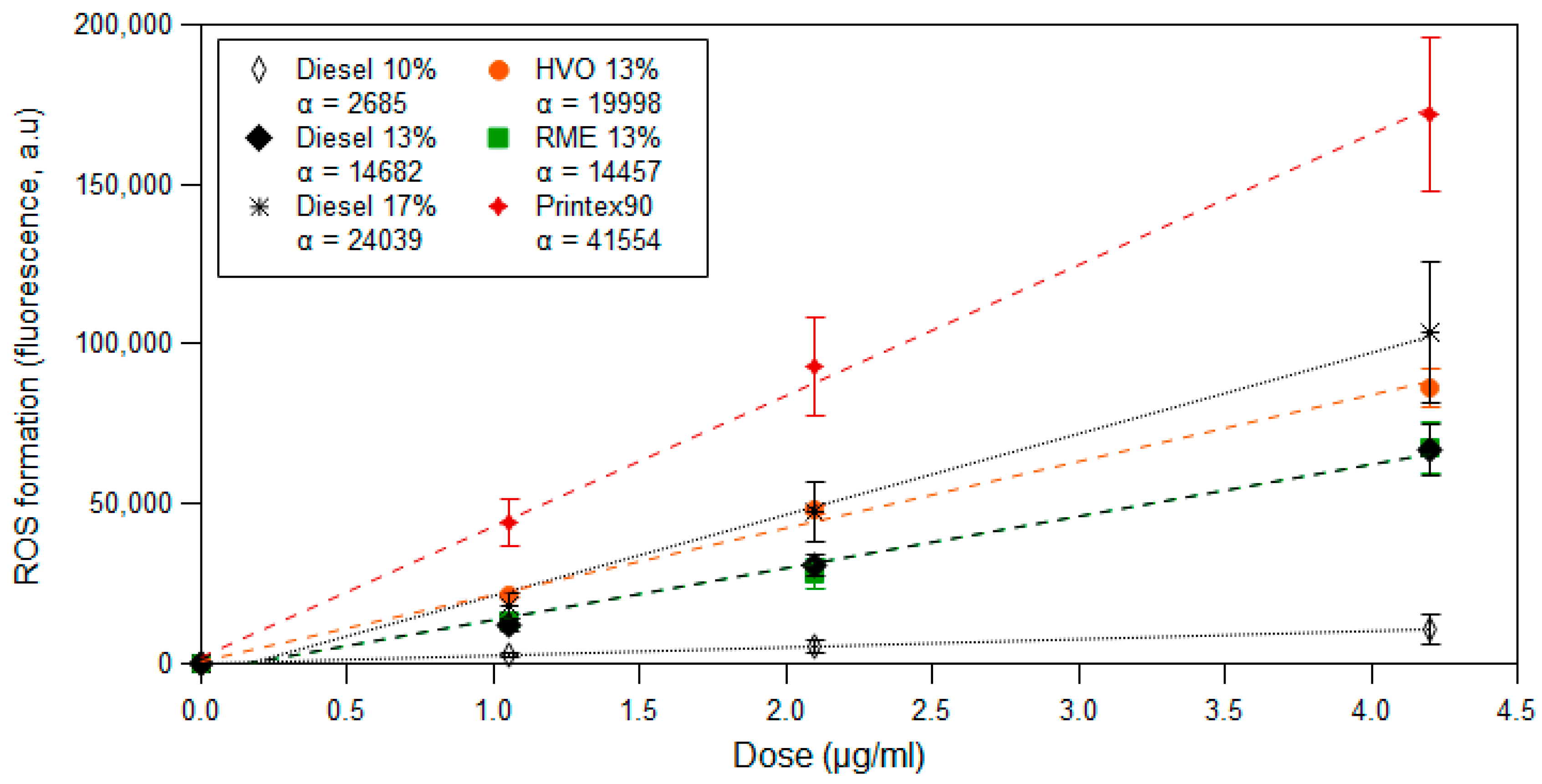
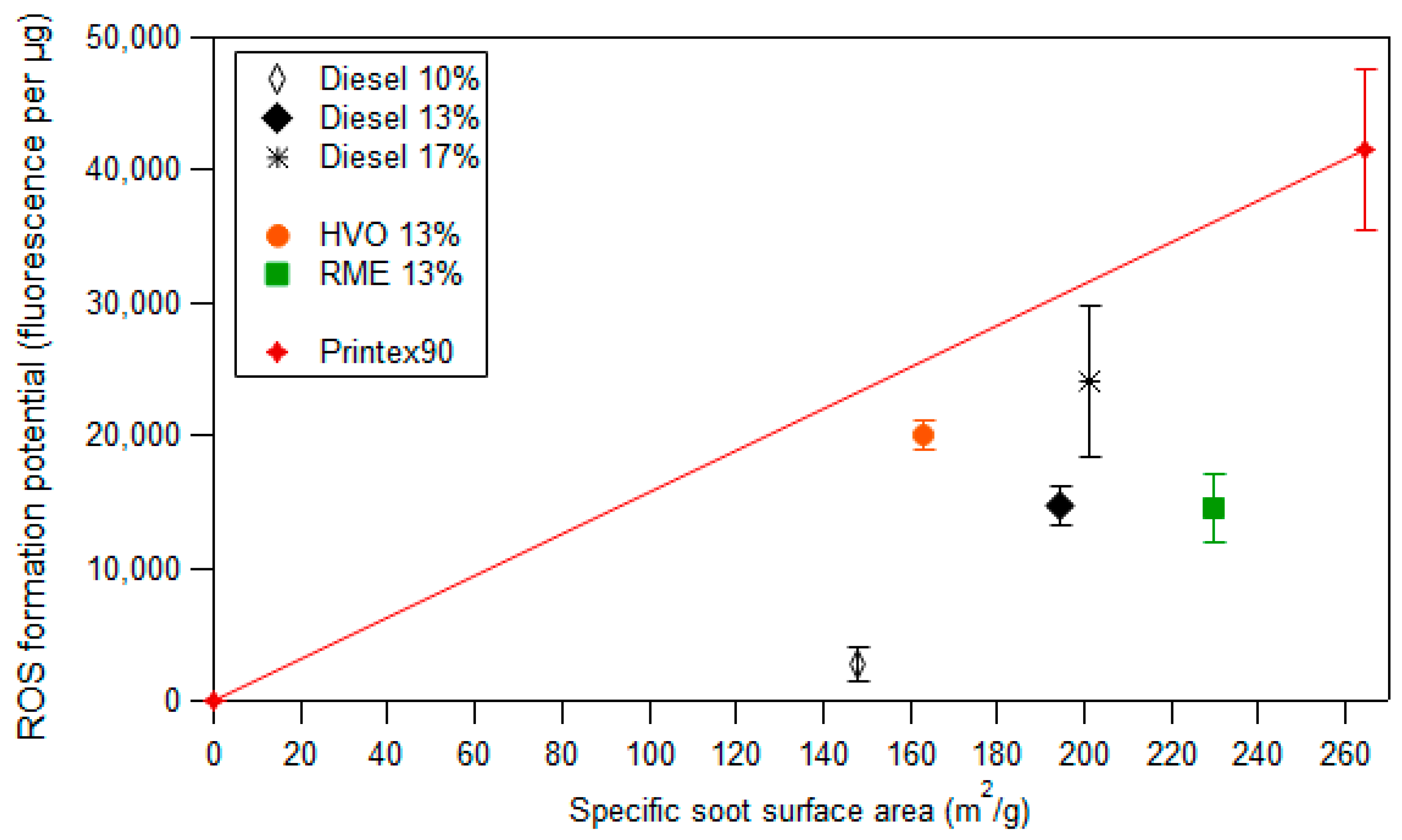
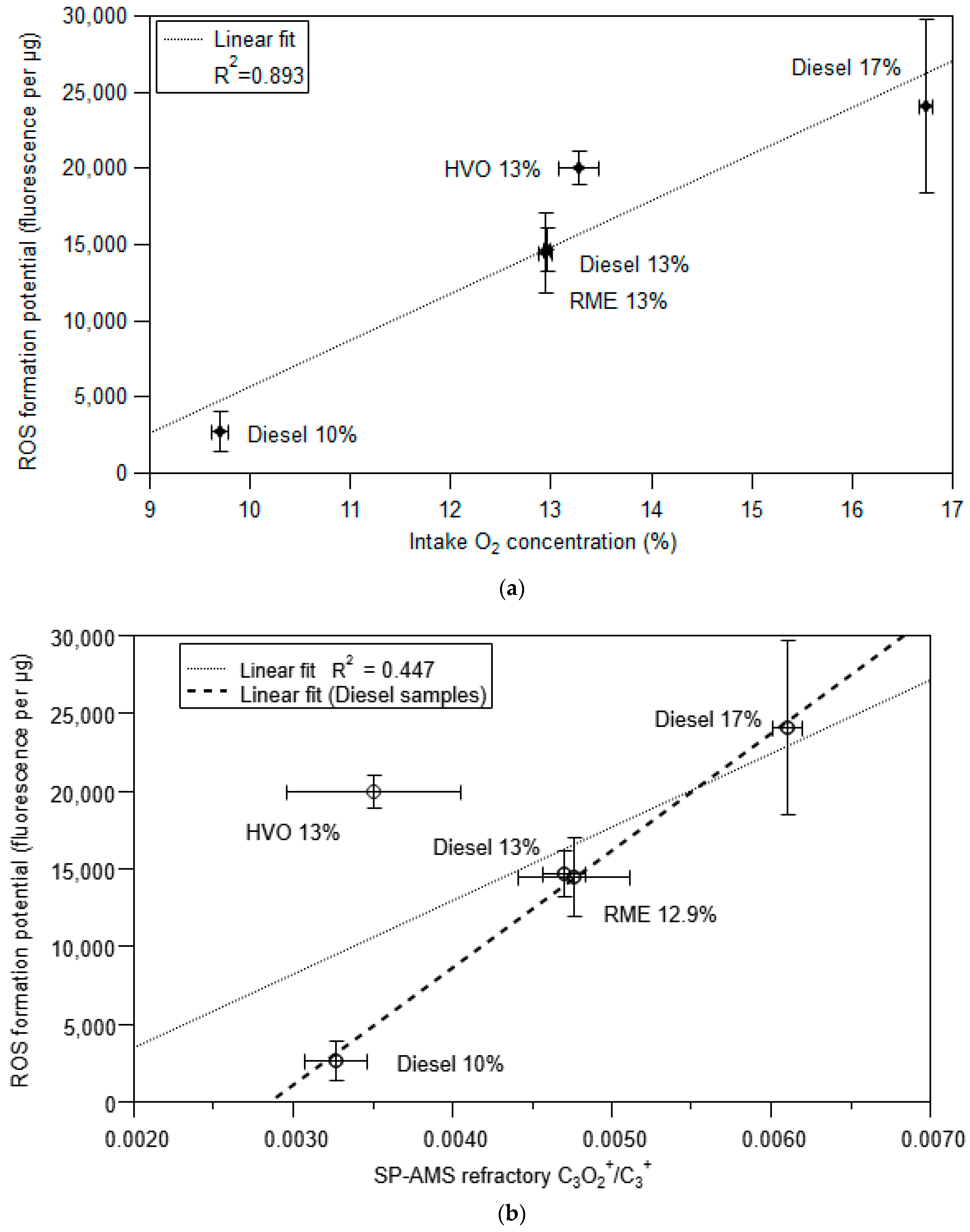
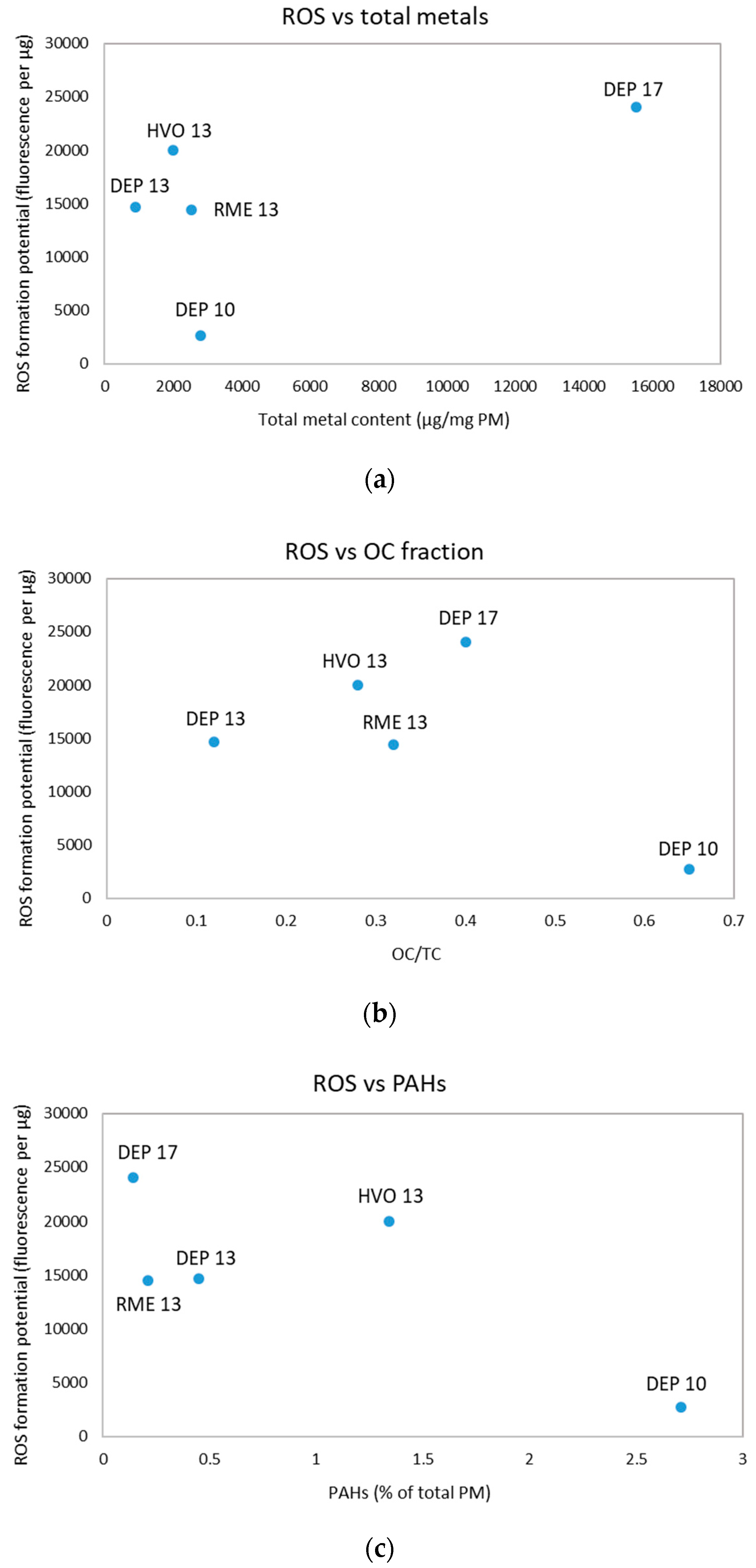
3.3. ROS Formation Potential of Diesel Exhaust Particles
3.4. Framework to Interpret Particle ROS Formation Potential Based on Fundamental Soot Formation and Oxidation Mechanisms in Combustion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Method

Appendix A.1.1. SP-AMS
Appendix A.1.2. PAH Analysis
Chemicals and Reagents
Sample Extraction, Clean-Up and Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | m/z | RT | IS | m/z | RT |
|---|---|---|---|---|---|
| naphthalene | 128 | 11.95 | naphthalene-d8 | 136 | 11.90 |
| 2-methylnaphthalene | 142 | 13.61 | acenaphthylene-d8 | 160 | 15.74 |
| 1-methylnaphthalene | 142 | 13.83 | acenaphthylene-d8 | 160 | 15.74 |
| biphenyl | 154 | 14.77 | acenaphthylene-d8 | 160 | 15.74 |
| 2,3-dimethylnaphthalene | 156 | 15.59 | acenaphthylene-d8 | 160 | 15.74 |
| acenaphthylene | 152 | 15.74 | acenaphthylene-d8 | 160 | 15.74 |
| acenaphthene | 154 | 16.18 | acenaphthene-d10 | 164 | 16.10 |
| 2,3,5-trimethylnaphthalene | 170 | 17.27 | acenaphthene-d10 | 164 | 16.10 |
| fluorene | 166 | 17.65 | fluorene-d10 | 176 | 17.56 |
| 1-methylfluorene | 180 | 19.92 | fluorene-d10 | 176 | 17.56 |
| phenanthrene | 178 | 21.88 | phenanthrene-d10 | 188 | 21.75 |
| anthracene | 178 | 22.17 | anthracene-d10 | 188 | 22.08 |
| 2-methylphenanthrene | 192 | 24.99 | phenanthrene-d10 | 188 | 21.75 |
| 1-methylphenantrene | 192 | 25.74 | phenanthrene-d10 | 188 | 21.75 |
| 1-methylanthracene | 192 | 25.82 | phenanthrene-d10 | 188 | 21.75 |
| 3-methylphenanthrene | 192 | 25.83 | phenanthrene-d10 | 188 | 21.75 |
| 2-phenylnaphthalene | 204 | 27.20 | phenanthrene-d10 | 188 | 21.75 |
| fluoranthene | 202 | 29.73 | fluoranthene-d10 | 212 | 29.61 |
| pyrene | 202 | 31.24 | pyrene-d10 | 212 | 31.12 |
| 1-methylfluoranthene | 216 | 33.83 | pyrene-d10 | 212 | 31.12 |
| retene | 234 | 34.06 | pyrene-d10 | 212 | 31.12 |
| 1-methylpyrene | 216 | 35.5 | pyrene-d10 | 212 | 31.12 |
| benzo(a)anthracene | 228 | 40.44 | benzo(a)anthracene-d12 | 240 | 40.29 |
| chrysene | 228 | 40.68 | chrysene-d12 | 240 | 40.49 |
| 2-methylchrysene | 228 | 40.68 | chrysene-d12 | 240 | 40.49 |
| benzo(b)fluoranthene | 264 | 47.96 | benzo(b)fluoranthene-d12 | 242 | 43.89 |
| benzo(k)fluoranthene | 264 | 48.19 | benzo(k)fluoranthene-d12 | 252 | 48.12 |
| benzo(a)pyrene | 252 | 50.14 | benzo(a)pyrene -d12 | 264 | 49.99 |
| perylene | 252 | 50.69 | benzo(a)pyrene-d12 | 264 | 264 |
| indeno(1,2,3-c,d)pyrene | 276 | 56.86 | indeno(1,2,3-c,d)pyrene-d12 | 288 | 57.03 |
| dibenzo(a,h)anthracene | 276 | 57.20 | dibenzo(a,h)anthracene-d14 | 288 | 57.03 |
| benzo(g,h,i)perylene | 276 | 58.19 | benzo(g,h,i)perylene-d12 | 288 | 58.05 |
| octachloronaphthalene (OCN, RS) | 404 | 49.38 |
| Name | m/z | RT | IS | m/z | RT |
|---|---|---|---|---|---|
| dibenzothiophene | 184 | 21.50 | phenanthrene-d10 | 188 | 21.75 |
| 2-methyldibenzothiophene | 198 | 24.21 | phenanthrene-d10 | 188 | 21.75 |
| 1-methyldibenzothiophene | 198 | 24.76 | phenanthrene-d10 | 188 | 21.75 |
| 4-methyldibenzothiophene | 198 | 25.38 | phenanthrene-d10 | 188 | 21.75 |
| 2,8-dimethyldibenzothiophene | 212 | 28.20 | fluoranthene-d10 | 212 | 29.61 |
| 2,4,7-trimethyldibenzothiophene | 226 | 30.95 | fluoranthene-d10 | 212 | 29.61 |
| Name | m/z | RT | IS | m/z | RT |
|---|---|---|---|---|---|
| nitro-PAHs | |||||
| 1-nitronaphthalene | 173 | 17.82 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 2-nitronaphthalene | 173 | 18.59 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 5-nitroacenaphthalene | 199 | 27.87 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 2-nitrofluorene | 211 | 30.81 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 9-nitroanthracene | 223 | 31.49 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 9-nitrophenanthrene | 223 | 33.65 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 3-nitrofluoranthene | 247 | 42.62 | 3-nitrofluoranthene-d9 | 256 | 42.70 |
| 4-nitropyrene | 247 | 42.98 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 1-nitropyrene | 247 | 43.99 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 2-nitropyrene | 247 | 44.54 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 7-nitrobenz(a)anthracene | 273 | 48.67 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 6-nitrochrysene | 273 | 50.72 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 3-nitronbenzanthrone | 275 | 50.88 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 1,3-dinitropyrene | 292 | 52.42 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 1,6-dinitropyrene | 292 | 53.71 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 1,8-dinitropyrene | 292 | 54.59 | 6-nitrochrysene-d9 | 284 | 50.70 |
| 6-Nitrobenzo(a)pyrene | 297 | 57.73 | 6-nitrochrysene-d9 | 284 | 50.70 |
| oxy-PAHs | |||||
| naphthalene-1-aldehyde | 156 | 16.38 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 2-naphthaldehyde | 156 | 16.42 | 2-nitrofluorene-d9 | 220 | 30.80 |
| p-fluorenone | 180 | 20.57 | 2-nitrofluorene-d9 | 220 | 30.80 |
| phenanthrene-9-aldehyde | 206 | 31.23 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 9,10-anthraquinone | 208 | 26.92 | 2-nitrofluorene-d9 | 220 | 30.80 |
| 1,4-anthraquinone | 208 | 29.01 | 2-nitrofluorene-d9 | 220 | 30.80 |
| benzo(a)fluorene | 230 | 37.57 | 3-nitrofluoranthene-d9 | 256 | 42.70 |
| benzo(b)fluorene | 230 | 39.21 | 3-nitrofluoranthene-d9 | 256 | 42.70 |
| benzanthrone | 230 | 41.34 | 3-nitrofluoranthene-d9 | 256 | 42.70 |
| benz(a)anthracene-7,12-dione | 258 | 43.73 | 3-nitrofluoranthene-d9 | 256 | 42.70 |
Appendix A.2. Results
Variability




| DEP | HVO | RME | Reference | |||
|---|---|---|---|---|---|---|
| MK1 Low-Sulfur Diesel | Hydrotreated Vegetable Oil | Rapeseed Methyl Ester | NIST2975 | |||
| 10 | 13 | 17 | 13 | 13 | ||
| Native PAH | ||||||
| naphthalene | 4.4 | 136.8 | 6.8 | 52.8 | 19.3 | 1.4 (1.8) |
| biphenyl | 5.0 | 67.7 | 2.7 | 63.2 | 3.5 | 0.3 (0.3) |
| acenaphthylene | 16.2 | 96.9 | 2.8 | 68.7 | 5.7 | <LOD |
| acenaphthene | 1.0 | 17.0 | 2.0 | 3.5 | 0.5 | 0.1 |
| fluorene | 17.5 | 44.8 | 2.9 | 62.5 | 5.1 | 0.5(0.4) |
| phenanthrene | 423.7 | 236.9 | 44.7 | 1167.4 | 207.8 | 10.6(17.3) |
| anthracene | 60.9 | 17.8 | 3.3 | 47.9 | 21.4 | 0.1(0.0) |
| fluoranthene | 1677.0 | 141.4 | 162.2 | 1591.4 | 325.2 | 23.3 (26.9) |
| pyrene | 2381.6 | 182.8 | 223.2 | 2341.4 | 293.1 | 0.8 (0.9) |
| retene | 9.1 | 271.8 | 166.1 | 120.6 | 157.6 | <LOD |
| benzo(a)anthracene | 1378.15 | 5.5 | 15.2 | 187.7 | 6.3 | 0.2 (0.3) |
| chrysene | 2609.2 | 9.3 | 52.8 | 369.9 | 15.8 | 3.9 (4.6) |
| benzo(b)fluoranthene | 4158.16 | 207.6 | 40.7 | 548.4 | 27.8 | 8.2(11.5) |
| benzo(k)fluoranthene | 1094. | 887.6 | 59.0 | 187.9 | 36.2 | 0.3 (0.7) |
| benzo(a)pyrene | 3739.3 | 61.3 | 32.8 | 759.2 | 33.4 | 0.3(0.0) |
| perylene | 638.3 | 14.5 | 10.0 | 137.6 | 2.5 | 0.1(0.0) |
| indeno(1,2,3-c,d)pyrene | 2072.1 | 2.3 | 8.7 | 702.5 | 2.1 | 0.9 (1.4) |
| dibenzo(a,h)anthracene | 40.2 | 27.2 | 7.1 | 49.9 | 4.3 | 0.4(0.3) |
| benzo(g,h,i)perylene | 2608.5 | 31.6 | 14.3 | 1216.6 | 7.6 | 0.9(0.5) |
| coronene | 752.3 | 7.6 | 0.5 | 280.5 | 0.8 | 0.3 |
| SUM Native PAH | 23,686.3 | 2468.7 | 857.6 | 9959.3 | 1176.1 | 52.4 |
| Alkyl-PAH | ||||||
| 2-methylnaphthalene | <LOD | 150.3 | 2.8 | 58.3 | 10.4 | 0.8(1.9) |
| 1-methylnaphthalene | 1.4 | 106.5 | 2.5 | 30.8 | 6.4 | 0.4(1.0) |
| 2,3-dimethylnaphthalene | 3.8 | 51.8 | 7.3 | 14.6 | 13.5 | 2.4 |
| 2,3,5-trimethylnaphthalene | 0.2 | 14.7 | 2.1 | 3.3 | 0.8 | 0.2 |
| 1-methylfluorene | 0.4 | 11.1 | 3.3 | 7.8 | 3.3 | 0.1 |
| 4-methylphenanthrene | 8.5 | 16.9 | 7.1 | 15.5 | 9.7 | 0.6 |
| 3-methylphenanthrene | 11.0 | 14.1 | 7.8 | 18.6 | 13.3 | 1.2 (1.0) |
| 1-methylphenanthrene | 11.5 | 16.6 | <LOD | 18.1 | 56.0 | <LOD |
| 1-methylanthracene | 17.6 | 16.6 | 6.9 | 23.3 | 11.9 | 0.6 |
| 2-phenylnaphthalene | 83.1 | 25.5 | 6.3 | 81.1 | 26.9 | 1.1 |
| 1-methylfluoranthene | 41.8 | 23.7 | 6.9 | 98.6 | 17.2 | 0.2 |
| 1-methylpyrene | 166.8 | 34.7 | 23.3 | 268.3 | 30.4 | 0.0 |
| 2-methylchrysene | 54.4 | 0.4 | 1.2 | 6.1 | 0.7 | 0.0 |
| SUM Alkyl-PAH | 400.4 | 482.9 | 77.2 | 644.3 | 150.5 | 7.5 |
| DBT (Dibenzothiophenes) | ||||||
| dibenzothiophene | 44.1 | 52.3 | 99.7 | 61.1 | 72.0 | 10.3 |
| 2-methyldibenzothiophene | 1.1 | 4.8 | 13.1 | 8.0 | 6.4 | 0.1 |
| 1-methyldibenzothiophene | 0.4 | 12.4 | 6.6 | 10.6 | 8.4 | 0.2 |
| 4-methyldibenzothiophene | 0.2 | 1.0 | 1.2 | 0.9 | 0.7 | 0.0 |
| 2,8-dimethyldibenzothiophene | 0.5 | 5.9 | 4.0 | 4.1 | 4.5 | 0.0 |
| 2,4,7-trimethyldibenzothiophene | 0.5 | 1.9 | 3.3 | 1.8 | 2.5 | <LOD |
| SUM DBT | 46.8 | 78.2 | 127.8 | 86.3 | 94.5 | 10.5 |
| Nitro-PAH | ||||||
| 1-Nitronaphthalene | 0.1 | 1.6 | 0.1 | 2.2 | 3.0 | 0.0(0.0) |
| 2-Nitronapthalene | 0.3 | 11.9 | 0.8 | 22.7 | 13.0 | 0.1(0.1) |
| 5-nitro acenapthalene | 1.3 | <LOD | 2.0 | 0.5 | <LOD | <LOD |
| 2-Nitrofluorene | 0.1 | 0.0 | 0.0 | 0.1 | 0.5 | 0.1 |
| 9-Nitroanthracene | 0.6 | 0.0 | 0.0 | 0.1 | <LOD | 1.6(3.0) |
| 9-Nitrophenanthrene | 0.7 | 0.0 | 0.0 | 0.2 | 0.0 | 0.2 |
| 4-Nitropyrene | 9.4 | 0.0 | 0.3 | 3.5 | 1.6 | 0.1(0.2) |
| 3-Nitrofluoranthene | 9.2 | 0.0 | <LOD | 1.2 | 2.1 | 2.2 |
| 1-Nitropyrene | 2.4 | 0.1 | 4.0 | 3.1 | 6.5 | 25.0 |
| 2-Nitropyrene | 88.6 | 0.2 | 0.2 | 26.2 | 1.8 | 0.0 |
| 7-Nitrobenz(a)anthracene | 7.0 | 6.4 | <LOD | 3.7 | 9.4 | 3.2 |
| 6-Nitrochrysene | 0.2 | <LOD | <LOD | 0.0 | 1.7 | 0.8 |
| 3-Nitrobenzanthrone | 0.3 | 0.1 | 0.2 | 0.7 | 0.3 | 0.2 |
| 6-Nitrobenzo(a)pyrene | 10.4 | 0.8 | 0.8 | 1.3 | 0.5 | 0.2(1.4) |
| SUM Nitro-PAHs | 130.5 | 21.2 | 8.4 | 65.5 | 40.4 | 33.7 |
| Oxy-PAH | ||||||
| Napthalene-1-aldehyde | 17.7 | 246.9 | 11.9 | 259.9 | 27.5 | 3.0 |
| 2-Naphthaldehyde | 67.6 | 1005.5 | 104.0 | 947.4 | 248.0 | 2.8 |
| p-Fluorenone | 136.0 | 168.3 | 49.7 | 659.8 | 214.5 | 12.7 |
| 9,10 Anthraquinone | 146.6 | 10.5 | 31.9 | 184.8 | 72.6 | 18.0 |
| 1,4 Anthraquinone | 25.2 | 6.5 | 1.3 | 2.7 | <LOD | <LOD |
| Phenanthrene-9-aldehyde | 50.3 | 0.5 | 2.8 | 21.7 | 0.1 | 2.0 |
| Benzanthrone | 1475.6 | 1.3 | 21.6 | 425.3 | 19.2 | 10.3 |
| Benz(a)anthracene-7,12-dione | 201.3 | 0.1 | 0.8 | 8.0 | 2.7 | 13.2 |
| SUM Oxy-PAH | 2488.3 | 1452.3 | 314.3 | 2627.5 | 596.3 | 265.1 |

References
- Agarwal, A.K.; Gupta, T.; Shukla, P.C.; Dhar, A. Particulate emissions from biodiesel fuelled CI engines. Energy Convers. Manag. 2015, 94, 311–330. [Google Scholar] [CrossRef]
- Lapuerta, M.; Octavio, A.; Rodriguez-Fernandez, J. Effect of biodiesel on diesel engine emissions. Prog. Energy Combust. Sci. 2008, 34, 198–223. [Google Scholar] [CrossRef]
- Murtonen, T.; Aakko-Saksa, P.; Kuronen, M.; Mikkonen, S.; Lehtoranta, K. Emissions with heavy-duty diesel engines and vehicles using FAME, HVO and GTL fuels with and without DOC + POC aftertreatment. SAE Int. J. Fuels Lubr. 2010, 2, 147–166. [Google Scholar] [CrossRef]
- Kuronen, M.; Mikkonen, S.; Aakko, P.; Murtonen, T. Hydrotreated Vegetable Oil as Fuel for Heavy Duty Diesel Engines. SAE Tech. Pap. Ser. 2007. [Google Scholar] [CrossRef]
- Deng, W.; Hu, Q.; Liu, T.; Wang, X.; Zhang, Y.; Song, W.; Sun, Y.; Bi, X.; Yu, J.; Yang, W.; et al. Primary particulate emissions and secondary organic aerosol (SOA) formation from idling diesel vehicle exhaust in China. Sci. Total Environ. 2017, 593–594, 462–469. [Google Scholar] [CrossRef]
- Lund, M.T.; Berntsen, T.K.; Heyes, C.; Klimont, Z.; Samset, B.H. Global and regional climate impacts of black carbon and co-emitted species from the on-road diesel sector. Atmos. Environ. 2014, 98, 50–58. [Google Scholar] [CrossRef]
- Ris, C.U.S. EPA Health assessment for diesel engine exhaust: A review. Inhal. Toxicol. 2007, 19, 229–239. [Google Scholar] [CrossRef]
- Hesterberg, T.W.; Long, C.M.; Bunn, W.B.; Lapin, C.A.; McClellan, R.O.; Valberg, P.A. Health effects research and regulation of diesel exhaust: An historical overview focused on lung cancer risk. Inhal. Toxicol. 2012, 24, 1–45. [Google Scholar] [CrossRef]
- IARC. Diesel and Gasoline Engine Exhausts and Some Nitroarenes; International Agency for Research on Cancer: Lyon, France, 2013; Volume 105. [Google Scholar]
- Brightwell, J.; Fouiliet, X.; Cassano-Zoppi, A.; Bernstein, D.; Crawley, F.; Duchosal, F.; Gatz, R.; Perczel, S.; Pfeifer, H. Tumours of the respiratory tract in rats and hamsters following chronic inhalation of engine exhaust emissions. J. Appl. Toxicol. 1989, 9, 23–31. [Google Scholar] [CrossRef]
- Li, N.; Xia, T.; Nel, A.E. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic. Biol. Med. 2008, 44, 1689–1699. [Google Scholar] [CrossRef]
- Nel, A.E.; Diaz-Sanchez, D.; Ng, D.; Hiura, T.; Saxon, A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J. Allergy Clin. Immunol. 1998, 102, 539–554. [Google Scholar] [CrossRef]
- Stoeger, T.; Reinhard, C.; Takenaka, S.; Schroeppel, A.; Karg, E.; Ritter, B.; Heyder, J.; Schulz, H. Instillation of six different ultrafine carbon particles indicates a surface area threshold dose for acute lung inflammation in mice. Environ. Health Perspect. 2006, 114, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.; Miller, M.R.; Clift, M.J.D.; Elder, A.; Mills, N.L.; Møller, P.; Schins, R.P.F.; Vogel, U.; Kreyling, W.G.; Jensen, K.A.; et al. Nanomaterials versus ambient ultrafine particles: An opportunity to exchange toxicology knowledge. Environ. Health Perspect. 2017, 125, 1–17. [Google Scholar] [CrossRef]
- Matti Maricq, M. Chemical characterization of particulate emissions from diesel engines: A review. J. Aerosol Sci. 2007, 38, 1079–1118. [Google Scholar] [CrossRef]
- Stoeger, T.; Takenaka, S.; Frankenberger, B.; Ritter, B.; Karg, E.; Maier, K.; Schulz, H.; Schmid, O. Deducing in vivo toxicity of combustion-derived nanoparticles from a cell-Free oxidative potency assay and Metabolic activation of organic compounds. Environ. Health Perspect. 2009, 117, 54–60. [Google Scholar] [CrossRef]
- Karavalakis, G.; Gysel, N.; Schmitz, D.A.; Cho, A.K.; Sioutas, C.; Schauer, J.J.; Cocker, D.R.; Durbin, T.D. Impact of biodiesel on regulated and unregulated emissions, and redox and proinflammatory properties of PM emitted from heavy-duty vehicles. Sci. Total Environ. 2017, 584–585, 1230–1238. [Google Scholar] [CrossRef]
- Stevanovic, S.; Miljevic, B.; Surawski, N.C.; Fairfull-Smith, K.E.; Bottle, S.E.; Brown, R.; Ristovski, Z.D. Influence of oxygenated organic aerosols (OOAs) on the oxidative potential of diesel and biodiesel particulate matter. Environ. Sci. Technol. 2013, 47, 7655–7662. [Google Scholar] [CrossRef]
- Jalava, P.I.; Aakko-Saksa, P.; Murtonen, T.; Happo, M.S.; Markkanen, A.; Yli-Pirilä, P.; Hakulinen, P.; Hillamo, R.; Mäki-Paakkanen, J.; Salonen, R.O.; et al. Toxicological properties of emission particles from heavy duty engines powered by conventional and bio-based diesel fuels and compressed natural gas. Part. Fibre Toxicol. 2012, 9, 37. [Google Scholar] [CrossRef]
- Cheung, K.L.; Polidori, A.; Ntziachristos, L.; Cassee, F.R.; Gerlofs, M. Chemical Characteristics and Oxidative Potential of Particulate Matter Emissions from Gasoline, Diesel, and Biodiesel Cars. Environ. Sci. Technol. 2009, 43, 6334–6340. [Google Scholar] [CrossRef]
- Hedayat, F.; Stevanovic, S.; Milic, A.; Miljevic, B.; Nabi, M.N.; Zare, A.; Bottle, S.E.; Brown, R.J.; Ristovski, Z.D. Influence of oxygen content of the certain types of biodiesels on particulate oxidative potential. Sci. Total Environ. 2016, 545–546, 381–388. [Google Scholar] [CrossRef]
- Gerlofs-Nijland, M.E.; Totlandsdal, A.I.; Tzamkiozis, T.; Leseman, D.L.A.C.; Samaras, Z.; Låg, M.; Schwarze, P.; Ntziachristos, L.; Cassee, F.R. Cell toxicity and oxidative potential of engine exhaust particles: Impact of using particulate filter or biodiesel fuel blend. Environ. Sci. Technol. 2013, 47, 5931–5938. [Google Scholar] [CrossRef] [PubMed]
- Aatola, H.; Larmi, M.; Sarjovaara, T.; Mikkonen, S. Hydrotreated Vegetable Oil (HVO) as a Renewable Diesel Fuel: Trade-off between NOx, Particulate Emission, and Fuel Consumption of a Heavy Duty Engine. SAE Int. J. Engines 2008, 1. [Google Scholar] [CrossRef]
- Knothe, G. Biodiesel and renewable diesel: A comparison. Prog. Energy Combust. Sci. 2010, 36, 364–373. [Google Scholar] [CrossRef]
- Hassaneen, A.; Munack, A.; Ruschel, Y.; Schroeder, O.; Krahl, J. Fuel economy and emission characteristics of Gas-to-Liquid (GTL) and Rapeseed Methyl Ester (RME) as alternative fuels for diesel engines. Fuel 2012, 97, 125–130. [Google Scholar] [CrossRef]
- Wan Ghazali, W.N.M.; Mamat, R.; Masjuki, H.H.; Najafi, G. Effects of biodiesel from different feedstocks on engine performance and emissions: A review. Renew. Sustain. Energy Rev. 2015, 51, 585–602. [Google Scholar] [CrossRef]
- Mofijur, M.; Atabani, A.E.; Masjuki, H.H.; Kalam, M.A.; Masum, B.M. A study on the effects of promising edible and non-edible biodiesel feedstocks on engine performance and emissions production: A comparative evaluation. Renew. Sustain. Energy Rev. 2013, 23, 391–404. [Google Scholar] [CrossRef]
- Kim, D.S.; Hanifzadeh, M.; Kumar, A. Trend of biodiesel feedstock and its impact on biodiesel emission characteristics. Environ. Prog. Sustain. Energy 2018, 37, 7–19. [Google Scholar] [CrossRef]
- Savic, N.; Rahman, M.M.; Miljevic, B.; Saathoff, H.; Naumann, K.H.; Leisner, T.; Riches, J.; Gupta, B.; Motta, N.; Ristovski, Z.D. Influence of biodiesel fuel composition on the morphology and microstructure of particles emitted from diesel engines. Carbon N. Y. 2016, 104, 179–189. [Google Scholar] [CrossRef]
- Qu, L.; Wang, Z.; Zhang, J. Influence of waste cooking oil biodiesel on oxidation reactivity and nanostructure of particulate matter from diesel engine. Fuel 2016, 181, 389–395. [Google Scholar] [CrossRef]
- Zheng, M.; Reader, G.T.; Hawley, J.G. Diesel engine exhaust gas recirculation—A review on advanced and novel concepts. Energy Convers. Manag. 2004, 45, 883–900. [Google Scholar] [CrossRef]
- Tsolakis, A.; Megaritis, A.; Wyszynski, M.L.; Theinnoi, K. Engine performance and emissions of a diesel engine operating on diesel-RME (rapeseed methyl ester) blends with EGR (exhaust gas recirculation). Energy 2007, 32, 2072–2080. [Google Scholar] [CrossRef]
- Maiboom, A.; Tauzia, X.; Hétet, J.F. Experimental study of various effects of exhaust gas recirculation (EGR) on combustion and emissions of an automotive direct injection diesel engine. Energy 2008, 33, 22–34. [Google Scholar] [CrossRef]
- Al-Qurashi, K.; Boehman, A.L. Impact of exhaust gas recirculation (EGR) on the oxidative reactivity of diesel engine soot. Combust. Flame 2008, 155, 675–695. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.; Guan, C.; Huang, Z. Oxidative reactivity of particles emitted from a diesel engine operating at light load with EGR. Aerosol Sci. Technol. 2015, 49, 1–10. [Google Scholar] [CrossRef]
- Jung, Y.; Bae, C. Immaturity of soot particles in exhaust gas for low temperature diesel combustion in a direct injection compression ignition engine. Fuel 2015, 161, 312–322. [Google Scholar] [CrossRef]
- Thangaraja, J.; Kannan, C. Effect of exhaust gas recirculation on advanced diesel combustion and alternate fuels—A review. Appl. Energy 2016, 180, 169–184. [Google Scholar] [CrossRef]
- Novakovica, M.; Shamun, S.; Malmborg, V.B.; Kling, K.I.; Kling, J.; Vogel, U.B.; Tunestal, P.; Pagels, J.; Tuner, M. Regulated Emissions and Detailed Particle Characterisation for Diesel and RME Biodiesel Fuel Combustion with Varying EGR in a Heavy-Duty Engine. SAE Tech. Pap. 2019. [Google Scholar] [CrossRef]
- Török, S.; Malmborg, V.B.; Simonsson, J.; Eriksson, A.; Martinsson, J.; Mannazhi, M.; Pagels, J.; Bengtsson, P.E. Investigation of the absorption Ångström exponent and its relation to physicochemical properties for mini-CAST soot. Aerosol Sci. Technol. 2018, 52, 757–767. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Zhou, Q.; Zhang, X.; Xing, W.; Wei, Y.; Hu, M.; Zhao, L.; Toriba, A.; Hayakawa, K.; et al. Size distribution of particulate polycyclic aromatic hydrocarbons in fresh combustion smoke and ambient air: A review. J. Environ. Sci. (China) 2020, 88, 370–384. [Google Scholar] [CrossRef]
- Shukla, P.C.; Shamun, S.; Gren, L.; Malmborg, V.; Pagels, J.; Tuner, M. Investigation of Particle Number Emission Characteristics in a Heavy-Duty Compression Ignition Engine Fueled with Hydrotreated Vegetable Oil (HVO). SAE Tech. Pap. 2018. [Google Scholar] [CrossRef]
- Van Thuijl, E.; Roos, C.; Beurskens, L. An Overview of Biofuel Technologies, Markets and Policies in Europe; Energy Research Centre of the Netherlands ECN: Petten, The Netherlands, 2003; pp. 1–64. [Google Scholar]
- Tschanz, F.; Amstutz, A.; Onder, C.H.; Guzzella, L. A real-time soot model for emission control of a diesel engine. IFAC Proc. Vol. 2010, 43, 222–227. [Google Scholar] [CrossRef]
- Heywood, J.B. Internal Combustion Engine Fundamentals, 1st ed.; McGraw-Hill: New York, NY, USA, 1988; ISBN 0-07-028637-X 978-0-07-028637-5. [Google Scholar]
- Cavalli, F.; Viana, M.; Yttri, K.E.; Genberg, J.; Putaud, J. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: The EUSAAR protocol. Atmos. Meas. Tech. 2010, 3, 79–89. [Google Scholar] [CrossRef]
- Rissler, J.; Messing, M.E.; Malik, A.I.; Nilsson, P.T.; Nordin, E.Z.; Bohgard, M.; Sanati, M.; Pagels, J.H. Effective density characterization of soot agglomerates from various sources and comparison to aggregation theory. Aerosol Sci. Technol. 2013, 47, 792–805. [Google Scholar] [CrossRef]
- Malmborg, V.B.; Eriksson, A.C.; Shen, M.; Nilsson, P.; Gallo, Y.; Waldheim, B.; Martinsson, J.; Andersson, O.; Pagels, J. Evolution of in-cylinder diesel engine soot and emission characteristics investigated with on-line aerosol mass spectrometry. Environ. Sci. Technol. 2017, 51, 1876–1885. [Google Scholar] [CrossRef]
- Onasch, T.B.; Trimborn, A.; Fortner, E.C.; Jayne, J.T.; Kok, G.L.; Davidovits, P.; Worsnop, D.R.; Trimborn, A.; Fortner, E.C.; Jayne, J.T.; et al. Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application. Aerosol Sci. Technol. 2012, 46, 804–817. [Google Scholar] [CrossRef]
- Ruusunen, J.; Tapanainen, M.; Sippula, O.; Jalava, P.I.; Lamberg, H.; Nuutinen, K.; Tissari, J.; Ihalainen, M.; Kuuspalo, K.; Mäki-Paakkanen, J.; et al. A novel particle sampling system for physico-chemical and toxicological characterization of emissions. Anal. Bioanal. Chem. 2011, 401, 3183–3195. [Google Scholar] [CrossRef] [PubMed]
- Høgsberg, T.; Jacobsen, N.R.; Clausen, P.A.; Serup, J. Black tattoo inks induce reactive oxygen species production correlating with aggregation of pigment nanoparticles and product brand but not with the polycyclic aromatic hydrocarbon content. Exp. Dermatol. 2013, 22, 464–469. [Google Scholar] [CrossRef]
- Jacobsen, N.R.; Pojana, G.; White, P.; Möller, P.; Cohn, C.A.; Korsholm, K.S.; Vogel, U.; Marcomini, A.; Loft, S.; Wallin, H. Genotoxicity, Cytotoxicity, and Reactive Oxygen Species Induced by Single-Walled Carbon Nanotubes and C60 Fullerenes in the FE1-MutaTM Mouse Lung Epithelial Cells. Environ. Mol. Mutagen. 2008, 49, 249–255. [Google Scholar] [CrossRef]
- Jørgensen, R.B.; Strandberg, B.; Sjaastad, A.K.; Johansen, A.; Svendsen, K. Simulated restaurant cook exposure to emissions of PAHs, mutagenic aldehydes, and particles from frying bacon. J. Occup. Environ. Hyg. 2013, 10, 122–131. [Google Scholar] [CrossRef]
- Bendtsen, K.M.; Brostrøm, A.; Koivisto, A.J.; Koponen, I.; Berthing, T.; Bertram, N.; Kling, K.I.; Dal Maso, M.; Kangasniemi, O.; Poikkimäki, M.; et al. Airport emission particles: Exposure characterization and toxicity following intratracheal instillation in mice. Part. Fibre Toxicol. 2019, 16, 1–23. [Google Scholar] [CrossRef]
- Gallo, Y.; Svensson, E.; Shen, M.Q.; Tunér, M.; Andersson, Ö.; Malmborg, V.B.; Pagels, J.; Simonsson, J.; Bengtsson, P.E.; Garcia, A. Investigation of late-cycle soot oxidation using laser extinction and in-cylinder gas sampling at varying inlet oxygen concentrations in diesel engines. Fuel 2017, 193, 308–314. [Google Scholar] [CrossRef]
- Aronsson, U.; Chartier, C.; Andersson, Ö.; Johansson, B.; Sjöholm, J.; Wellander, R.; Richter, M.; Alden, M.; Miles, P. Analysis of EGR Effects on the Soot Distribution in a Heavy Duty Diesel Engine using Time-Resolved Laser Induced Incandescence. SAE Int. J. Engines 2010, 3, 137–155. [Google Scholar] [CrossRef]
- Sugiyama, K.; Goto, I.; Kitano, K.; Mogi, K.; Honkanen, M. Effects of hydrotreated vegetable Oil (HVO) as renewable diesel fuel on combustion and exhaust emissions in diesel engine. SAE Tech. Pap. 2011, 5, 205–217. [Google Scholar] [CrossRef]
- Dimitriadis, A.; Natsios, I.; Dimaratos, A.; Katsaounis, D. Evaluation of a Hydrotreated Vegetable Oil (HVO) and Effects on Emissions of a Passenger Car Diesel Engine. Front. Mech. Eng. 2018, 4, 1–19. [Google Scholar] [CrossRef]
- Bhardwaj, O.P.; Lüers, B.; Holderbaum, B.; Koerfer, T.; Pischinger, S.; Honkanen, M. Utilization of HVO Fuel Properties in a High Efficiency Combustion System: Part 2: Relationship of Soot Characteristics with its Oxidation Behavior in DPF. SAE Int. J. Fuels Lubr. 2014, 7, 979–994. [Google Scholar] [CrossRef]
- Prokopowicz, A.; Zaciera, M.; Sobczak, A.; Bielaczyc, P.; Woodburn, J. The Effects of Neat Biodiesel and Biodiesel and HVO Blends in Diesel Fuel on Exhaust Emissions from a Light Duty Vehicle with a Diesel Engine. Environ. Sci. Technol. 2015, 49, 7473–7482. [Google Scholar] [CrossRef]
- Malmborg, V.B.; Eriksson, A.C.; Török, S.; Zhang, Y.; Kling, K.; Martinsson, J.; Fortner, E.C.; Gren, L.; Kook, S.; Onasch, T.B.; et al. Relating aerosol mass spectra to composition and nanostructure of soot particles. Carbon N. Y. 2019, 142, 535–546. [Google Scholar] [CrossRef]
- Johansson, K.O.; El Gabaly, F.; Schrader, P.E.; Campbell, M.F.; Michelsen, H.A. Evolution of maturity levels of the particle surface and bulk during soot growth and oxidation in a flame. Aerosol Sci. Technol. 2017, 51, 1333–1344. [Google Scholar] [CrossRef]
- Chung, M.Y.; Lazaro, R.A.; Lim, D.; Jackson, J.; Lyon, J.; Rendulic, D.; Hasson, A.S. Aerosol-borne quinones and reactive oxygen species generation by particulate matter extracts. Environ. Sci. Technol. 2006, 40, 4880–4886. [Google Scholar] [CrossRef]
- Bobba, M.K.; Musculus, M.P.B. Laser diagnostics of soot precursors in a heavy-duty diesel engine at low-temperature combustion conditions. Combust. Flame 2012, 159, 832–843. [Google Scholar] [CrossRef]
- Shamun, S.; Novakovic, M.; Malmborg, V.B.; Preger, C.; Shen, M.; Messing, M.E.; Pagels, J.; Tunér, M.; Tunestäl, P. Detailed Characterization of Particulate Matter in Alcohol Exhaust Emissions. In Proceedings of the COMODIA 2017—9th International Conference on Modeling and Diagnostics for Advanved Engine Systems, Okayama, Japan, 25–28 July 2017. [Google Scholar]
- Jacobsen, N.R.; Møller, P.; Cohn, C.A.; Loft, S.; Vogel, U.; Wallin, H. Diesel exhaust particles are mutagenic in FE1-MutaTM Mouse lung epithelial cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2008, 641, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Schmid, O.; Stoeger, T. Surface area is the biologically most effective dose metric for acute nanoparticle toxicity in the lung. J. Aerosol Sci. 2016, 99, 133–143. [Google Scholar] [CrossRef]
- Monteiller, C.; Tran, L.; MacNee, W.; Faux, S.; Jones, A.; Miller, B.; Donaldson, K. The pro-inflammatory effects of low-toxicity low-solubility particles, nanoparticles and fine particles, on epithelial cells in vitro: The role of surface area. Occup. Environ. Med. 2007, 64, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Suárez, G.; Tran, N.; Puntes, V.; Riediker, M. Coating aerosolized nanoparticles with low-volatile organic compound (LVOC) vapors modifies surface functionality and oxidative reactivity. NanoImpact 2019, 14, 100150. [Google Scholar] [CrossRef]
- Zhou, J.; Elser, M.; Huang, R.-J.; Krapf, M.; Fröhlich, R.; Bhattu, D.; Stefenelli, G.; Zotter, P.; Bruns, E.A.; Pieber, S.; et al. Predominance of Secondary Organic Aerosol to Particle-bound Reactive Oxygen Species Activity in Fine Ambient Aerosol. Atmos. Chem. Phys. 2019, 19, 14703–14720. [Google Scholar] [CrossRef]
- Lee, J.C.; Son, Y.O.; Pratheeshkumar, P.; Shi, X. Oxidative stress and metal carcinogenesis. Free Radic. Biol. Med. 2012, 53, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.R.; Clausen, P.A. Carbon black nanoparticles and other problematic constituents of black ink and their potential to harm tattooed humans. Curr. Probl. Dermatol. 2015, 48, 170–175. [Google Scholar]
- Biswas, S.; Verma, V.; Schauer, J.J.; Cassee, F.R.; Cho, A.K.; Sioutas, C. Oxidative potential of semi-volatile and non volatile particulate matter (PM) from heavy-duty vehicles retrofitted with emission control technologies. Environ. Sci. Technol. 2009, 43, 3905–3912. [Google Scholar] [CrossRef]
- Khosousi, A.; Dworkin, S.B. Detailed modelling of soot oxidation by O2and OH in laminar diffusion flames. Proc. Combust. Inst. 2015, 35, 1903–1910. [Google Scholar] [CrossRef]
- Wang, L.; Song, C.; Song, J.; Lv, G.; Pang, H.; Zhang, W. Aliphatic C-H and oxygenated surface functional groups of diesel in-cylinder soot: Characterizations and impact on soot oxidation behavior. Proc. Combust. Inst. 2013, 34, 3099–3106. [Google Scholar] [CrossRef]
- Vander Wal, R.L.; Bryg, V.M.; Hays, M.D. Fingerprinting soot (towards source identification): Physical structure and chemical composition. J. Aerosol Sci. 2010, 41, 108–117. [Google Scholar] [CrossRef]
- Kholghy, M.R.; Veshkini, A.; Thomson, M.J. The core-shell internal nanostructure of soot—A criterion to model soot maturity. Carbon N. Y. 2016, 100, 508–536. [Google Scholar] [CrossRef]
- Corbin, J.C.; Sierau, B.; Gysel, M.; Laborde, M.; Keller, A.; Kim, J.; Petzold, A.; Onasch, T.B.; Lohmann, U.; Mensah, A.A. Mass spectrometry of refractory black carbon particles from six sources: Carbon-cluster and oxygenated ions. Atmos. Chem. Phys. 2014, 14, 2591–2603. [Google Scholar] [CrossRef]
- Herring, C.L.; Faiola, C.L.; Massoli, P.; Sueper, D.; Erickson, M.H.; McDonald, J.D.; Simpson, C.D.; Yost, M.G.; Jobson, B.T.; Van Reken, T.M. New Methodology for Quantifying Polycyclic Aromatic Hydrocarbons (PAHs) Using High-Resolution Aerosol Mass Spectrometry. Aerosol Sci. Technol. 2015, 49, 1131–1148. [Google Scholar] [CrossRef]








| Diesel | HVO | RME | ||||
|---|---|---|---|---|---|---|
| Average intake O2 concentration (% ± 1 std. dev.) | 9.7 ± 0.1 | 10.8 ± 0.5 | 13.1 ± 0.2 | 16.7 ± 0.4 | 13.3 ± 0.2 | 12.8 ± 0.5 |
| Equivalence ratio ± 1 std. dev. | 0.80 ± 0.00 | 0.73 ± 0.03 | 0.60 ± 0.00 | 0.43 ± 0.02 | 0.59 ± 0.01 | 0.65 ± 0.03 |
| Number of replicates | 3 (3) | 3 (0) | 6 (2) | 3 (3) | 2 (2) | 5 (2) |
| Calorific value (MJ/kg) | 43.15 | 44.1 * | 37.3 ** | |||
| Stoichiometric ratio | ~14.49 | ~14.90 | ~12.3 ** | |||
| Diesel | HVO | RME | Printex90 | ||||
|---|---|---|---|---|---|---|---|
| Average Intake O2 Concentration (%) | 10 | 12 | 13 | 17 | 13 | 13 | - |
| Primary particle size (GMD; nm) | 22.0 | 18.2 | 17.4 | 16.1 | 20.9 | 15.0 | 14.5 |
| (20.7, 23.4) | (16.3, 20.3) | (16.2, 18.8) | (14.9, 17.5) | (19.1, 22.8) | (13.7, 16.4) | (13.7, 15.4) | |
| Estimated SSA (m2/g) | 152 | 183 | 191 | 207 | 160 | 222 | 230 |
| (143, 161) | (164, 204) | (177, 206) | (191, 224) | (146, 174) | (203, 243) | (217, 243) | |
| Aggregate size (GMD; nm) from DMS | 55 ± 9 | 90 ± 5 | 104 ±8 | 62 ± 4 | 90 ± 5 | 70 ± 3 | - |
| Average number emissions (#/g PM) | 1.3 × 1016 | - | 3.6 × 1015 | 1.1 × 1016 | 6.3 × 1015 | 8.8 × 1015 | - |
| Diesel | HVO | RME | |||
|---|---|---|---|---|---|
| Average Intake O2 Concentration (%) | 10 | 13 | 17 | 13 | 13 |
| PM (gravimetric; mg/kWh) | 64 ± 11 | 386 ± 61 | 8 ± 2 | 133 ± 15 | 144 ± 27 |
| OC/TC | 0.65 | 0.12 | 0.40 | 0.28 | 0.32 |
| PAHs * (µg/g) | |||||
| Native PAHs | 23,700 | 2470 | 858 | 9960 | 1180 |
| Alkyl-PAHs | 400 | 483 | 77 | 644 | 150 |
| DBTs | 47 | 78 | 128 | 86 | 94 |
| Oxy-PAHs | 2490 | 1450 | 314 | 2630 | 596 |
| Nitro-PAHs | 131 | 21 | 8 | 65 | 40 |
| Total all PAHs | 26,800 | 4500 | 1390 | 13,400 | 2060 |
| Total all PAHs (µg/kWh) | 1514 | 954 | 7 | 1330 | 170 |
| Sum BaPeq (µg/g) | 4685 | 165 | 59 | 1067 | 60 |
| Metals (2 most abundant) | |||||
| Fe (µg/g) | 220 | 137 | 2120 | 247 | 116 |
| Cu (µg/g) | 2350 | 629 | 13,200 | 1630 | 2290 |
| Total all metals (µg/g) | 2820 | 905 | 15,500 | 1990 | 2530 |
| Total all metals (µg/kWh) | 180 | 350 | 121 | 266 | 364 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gren, L.; Malmborg, V.B.; Jacobsen, N.R.; Shukla, P.C.; Bendtsen, K.M.; Eriksson, A.C.; Essig, Y.J.; Krais, A.M.; Loeschner, K.; Shamun, S.; et al. Effect of Renewable Fuels and Intake O2 Concentration on Diesel Engine Emission Characteristics and Reactive Oxygen Species (ROS) Formation. Atmosphere 2020, 11, 641. https://doi.org/10.3390/atmos11060641
Gren L, Malmborg VB, Jacobsen NR, Shukla PC, Bendtsen KM, Eriksson AC, Essig YJ, Krais AM, Loeschner K, Shamun S, et al. Effect of Renewable Fuels and Intake O2 Concentration on Diesel Engine Emission Characteristics and Reactive Oxygen Species (ROS) Formation. Atmosphere. 2020; 11(6):641. https://doi.org/10.3390/atmos11060641
Chicago/Turabian StyleGren, Louise, Vilhelm B. Malmborg, Nicklas R. Jacobsen, Pravesh C. Shukla, Katja M. Bendtsen, Axel C. Eriksson, Yona J. Essig, Annette M. Krais, Katrin Loeschner, Sam Shamun, and et al. 2020. "Effect of Renewable Fuels and Intake O2 Concentration on Diesel Engine Emission Characteristics and Reactive Oxygen Species (ROS) Formation" Atmosphere 11, no. 6: 641. https://doi.org/10.3390/atmos11060641
APA StyleGren, L., Malmborg, V. B., Jacobsen, N. R., Shukla, P. C., Bendtsen, K. M., Eriksson, A. C., Essig, Y. J., Krais, A. M., Loeschner, K., Shamun, S., Strandberg, B., Tunér, M., Vogel, U., & Pagels, J. (2020). Effect of Renewable Fuels and Intake O2 Concentration on Diesel Engine Emission Characteristics and Reactive Oxygen Species (ROS) Formation. Atmosphere, 11(6), 641. https://doi.org/10.3390/atmos11060641