Characterization of Total OH Reactivity in a Rapeseed Field: Results from the COV3ER Experiment in April 2017

 ,
,
Abstract
1. Introduction
2. Experiments
2.1. Field Site Description
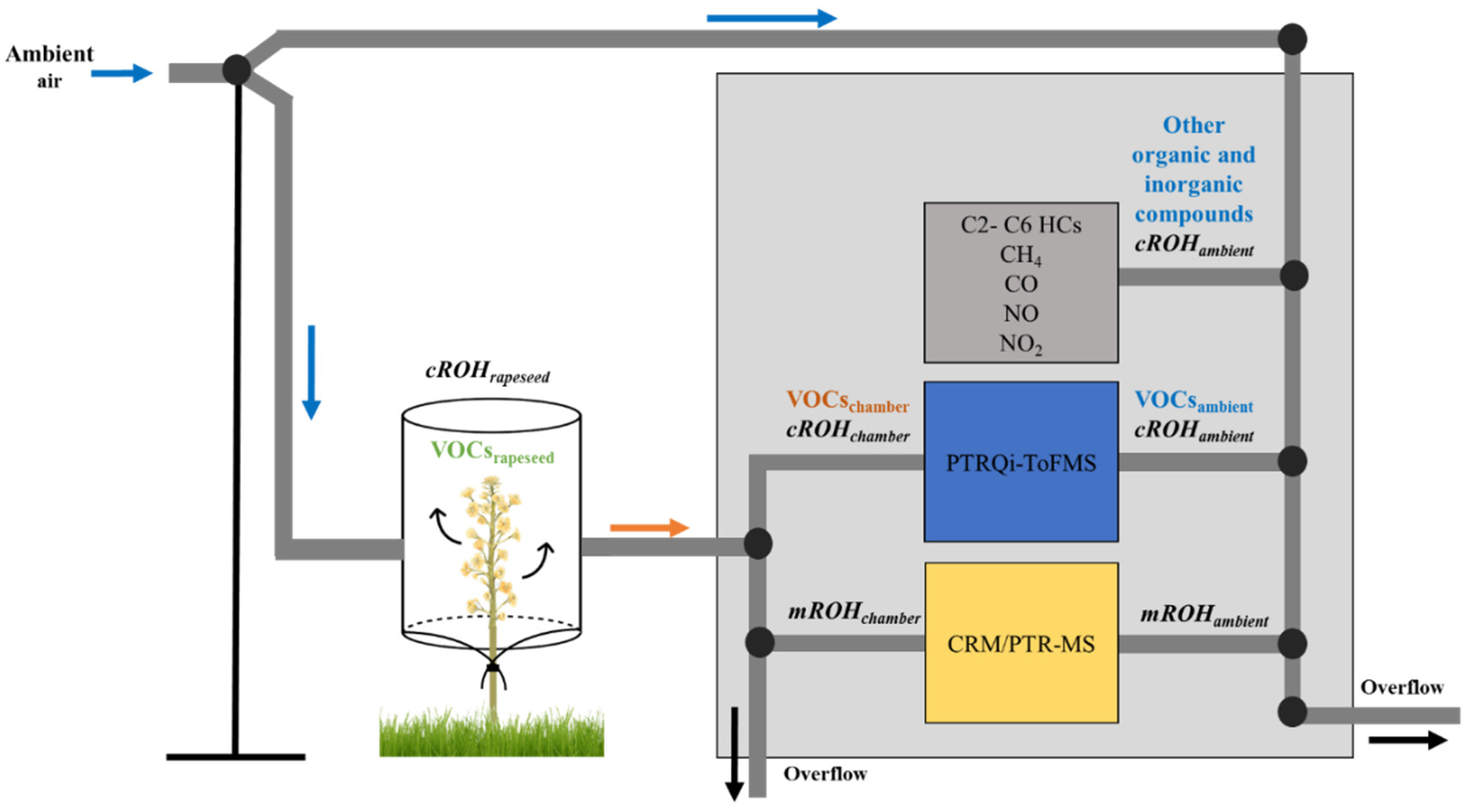
2.2. Plant Dynamic Chamber and Air Sampling
2.3. Meteorological Measurements
2.4. Instrumentation
2.4.1. Comparative Reactivity Method (CRM) and Instrument Performance
2.4.2. Proton Transfer Reaction Quadrupole ion-Time of Flight Mass Spectrometer (PTRQi-ToFMS)
2.4.3. Gas Chromatography-Flame Ionization Detector (GC- FID)
2.4.4. NOx Concentration Measurements
2.4.5. CO and CH4 Measurements
2.5. Calculation of OH Reactivity
2.6. Data Availability
3. Results and Discussion
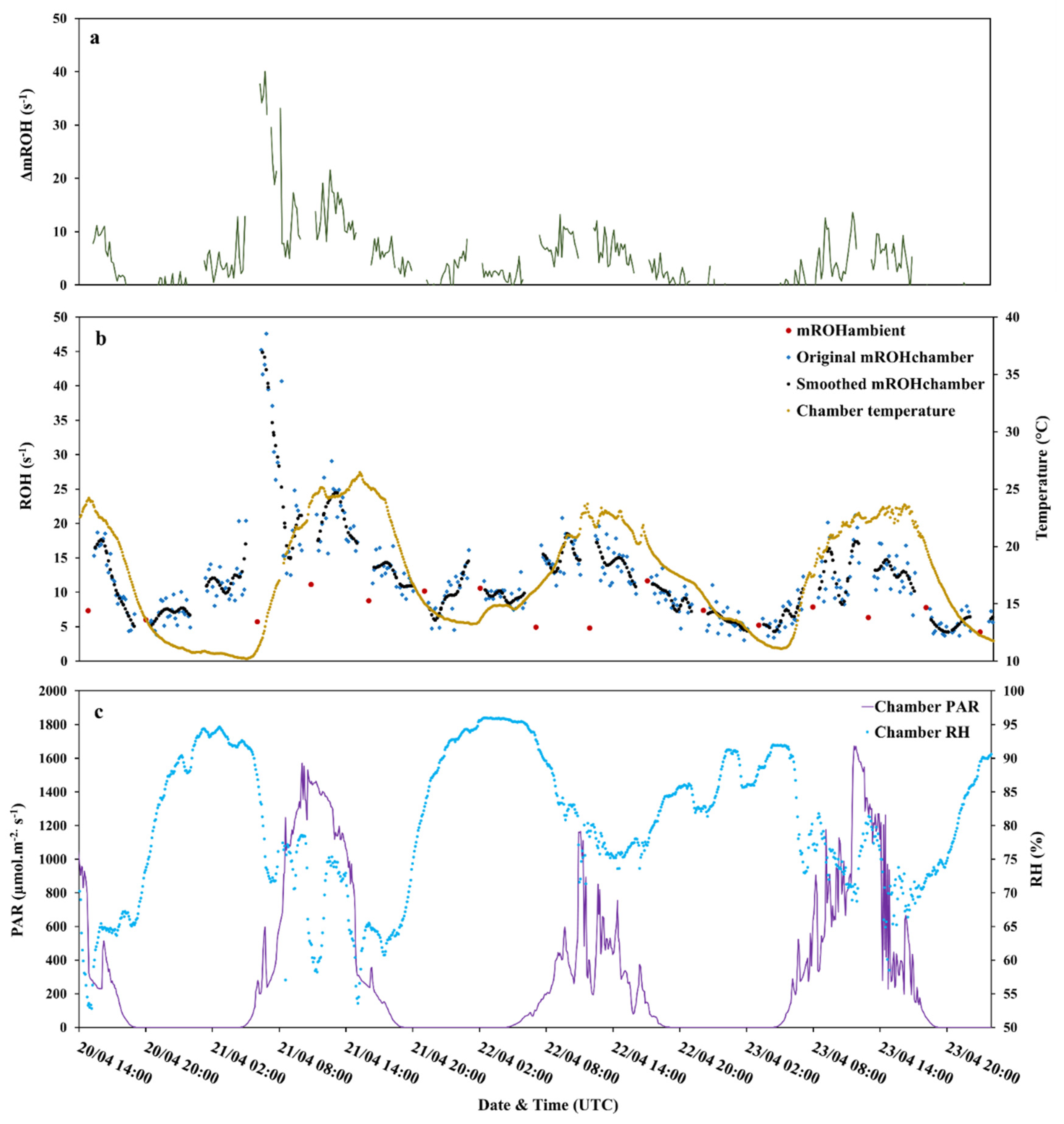
3.1. Total OH Reactivity
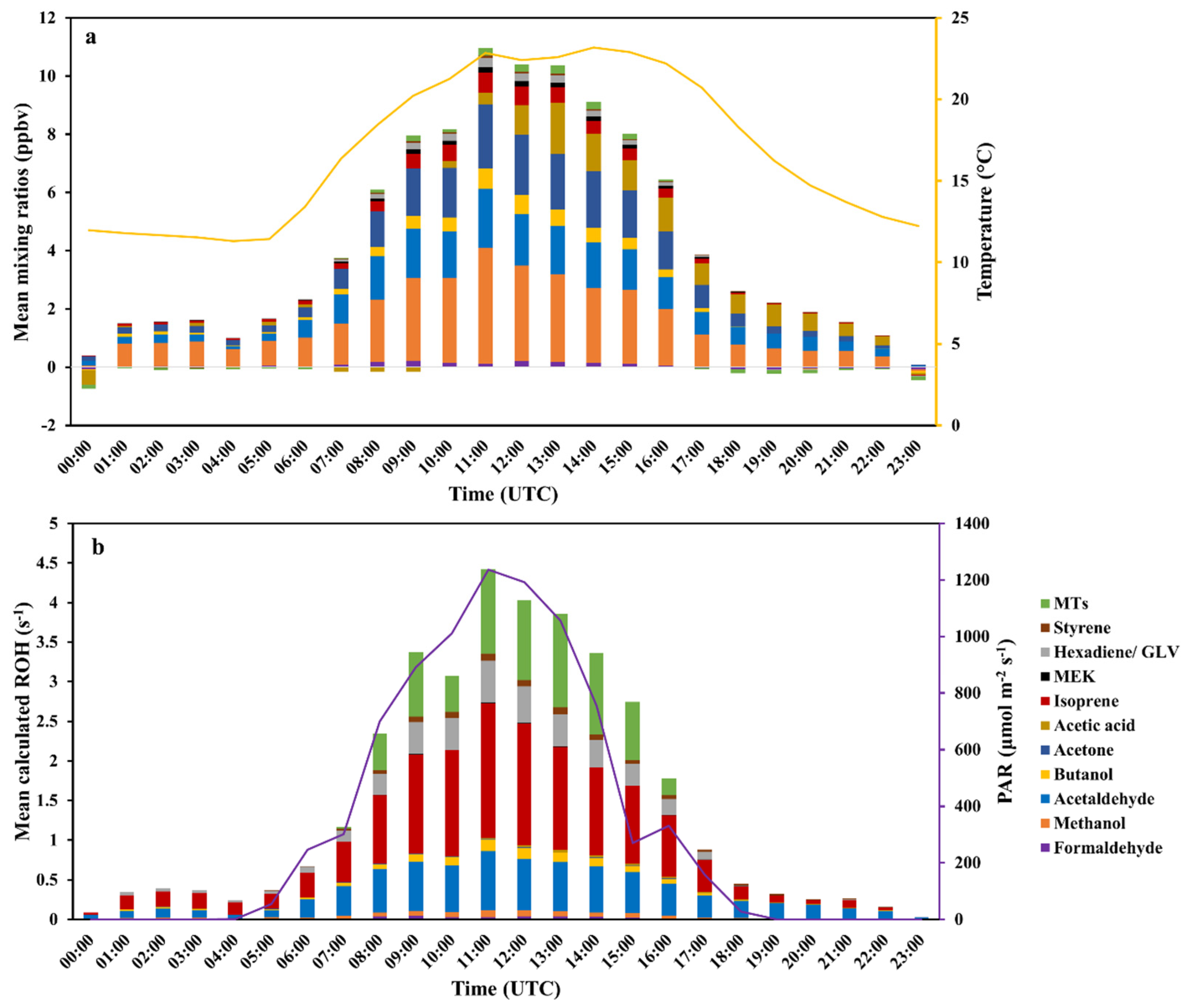
3.1.1. Ambient Air Measurements and their Calculated OH Reactivity (cROHambient)
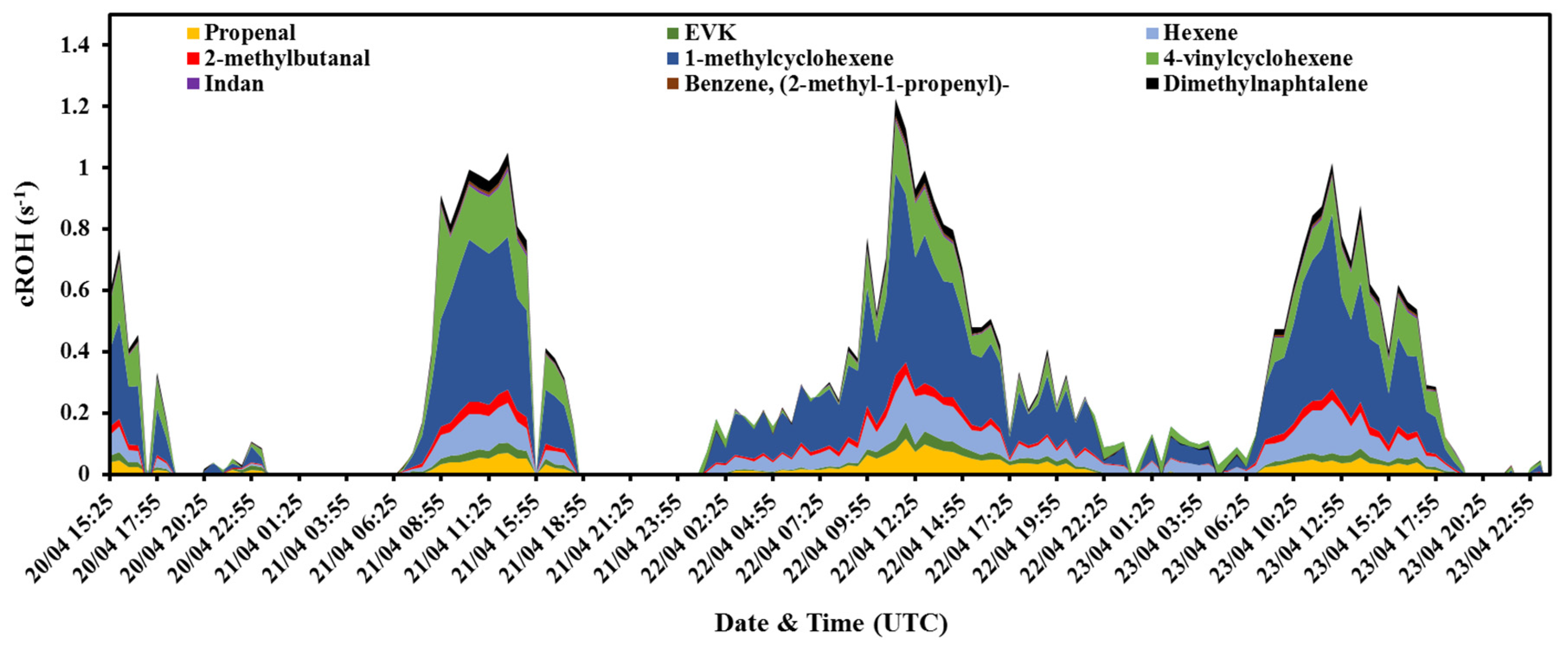
3.1.2. BVOCs from the Rapeseed Plant (VOCsrapeseed) and Their Calculated OH Reactivity (cROHrapeseed)
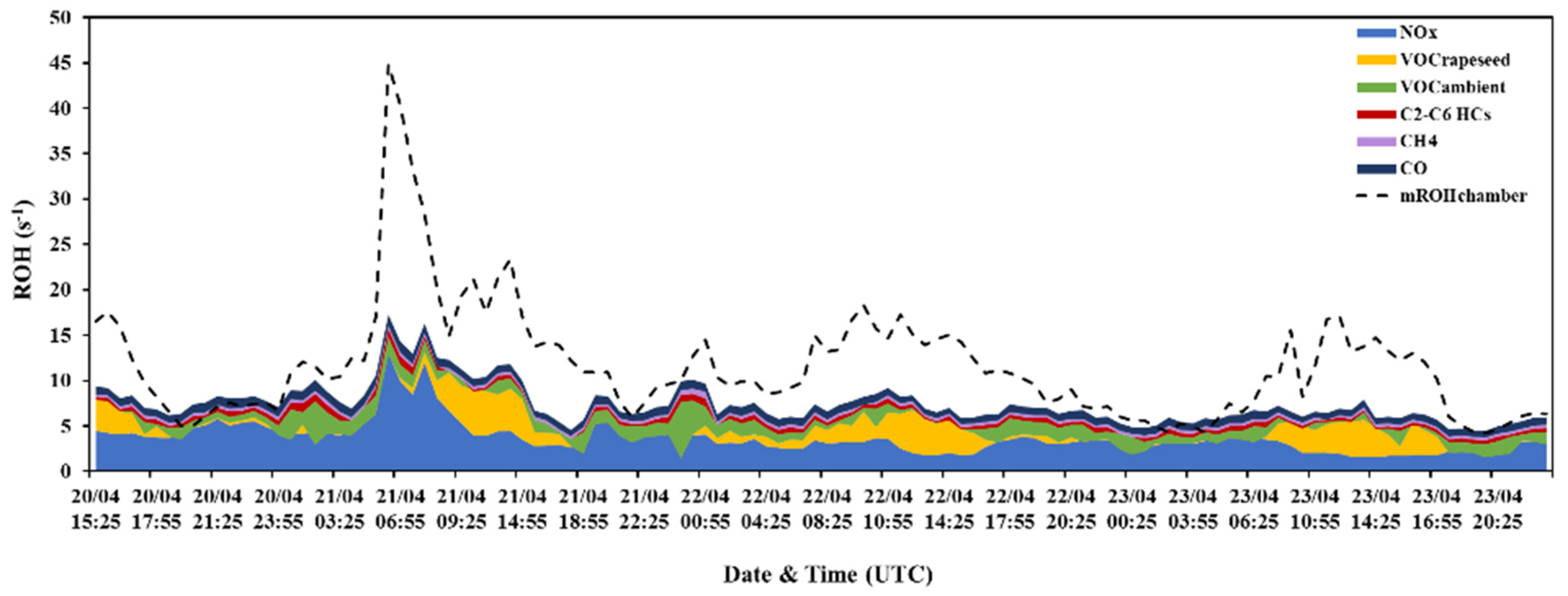
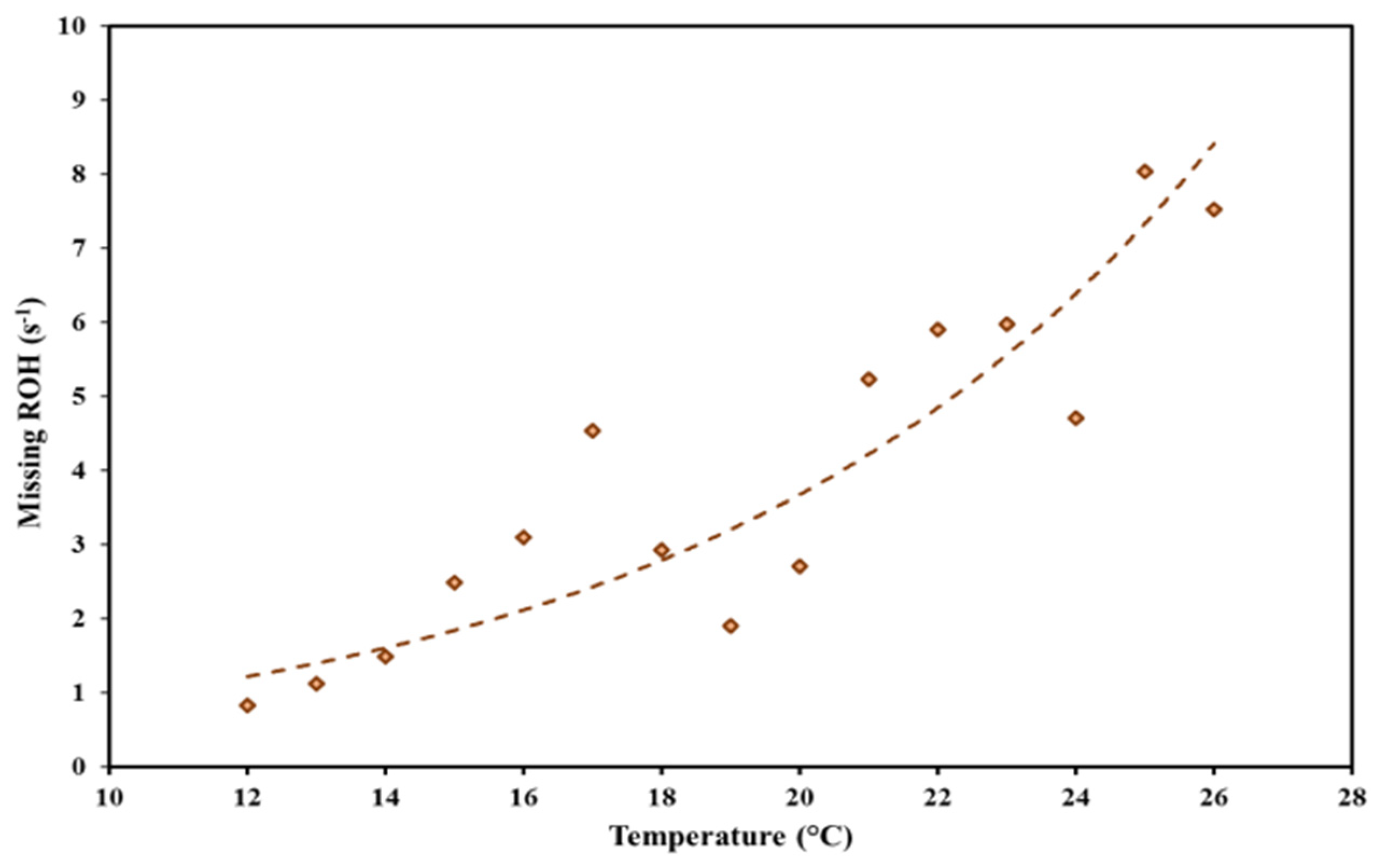
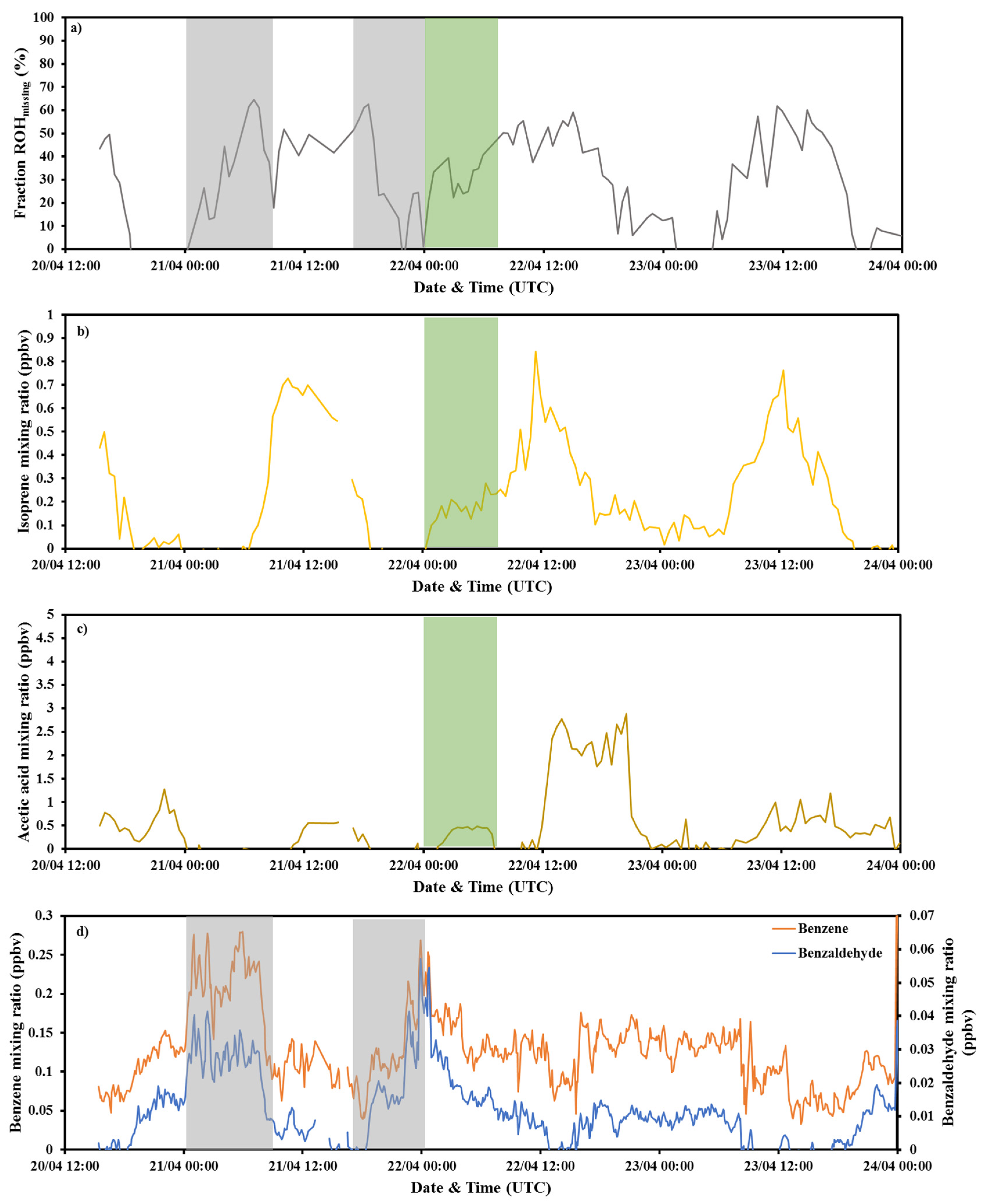
3.2. Investigation of the Missing OH Reactivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Goldstein, A.H.; Galbally, I.E. Known and unexplored organic constituents in the earth’s atmosphere. Environ. Sci. Technol. 2007, 41, 1514–1521. [Google Scholar] [CrossRef]
- Guenther, A. A global model of natural volatile organic compound emissions. J. Geophys. Res. 1995, 100, 8873–8892. [Google Scholar] [CrossRef]
- Helmig, D.; Bottenheim, J.; Galbally, I.E.; Lewis, A.; Milton, M.J.; Penkett, S.; Plass-Duelmer, C.; Reimann, S.; Tans, P.; Thiel, S. Volatile Organic Compounds in the Global Atmosphere. Eos (Washington. DC) 2009, 90, 513–514. [Google Scholar] [CrossRef]
- De Gouw, J.; Warneke, C. Measurements of volatile organic compounds in the earth’s atmosphere using proton-transfer-reaction mass spectrometry joost. Reprod. Hum. Horm. 2007, 26, 223–257. [Google Scholar]
- Laothawornkitkul, J.; Taylor, J.E.; Paul, N.D.; Hewitt, C.N. Biogenic volatile organic compounds in the Earth system: Tansley review. New Phytol. 2009, 183, 27–51. [Google Scholar] [CrossRef]
- Seigler, D.S. Chemistry and mechanisms of allelopathic interactions. Agron. J. 1996, 88, 876–886. [Google Scholar] [CrossRef]
- Kesselmeier, J.; Staudt, M. Biogenic volatile organic compounds (VOC): An overview on emission, physiology and ecology. J. Atmos. Chem. 1999, 33, 23–88. [Google Scholar] [CrossRef]
- Penuelas, J.; Llusia, J. The complexity of factors driving volatile organic compound emissions by plants. Biol. Plant. 2001, 44, 481–487. [Google Scholar] [CrossRef]
- Guenther, A. Biological and chemical diversity of biogenic volatile organic emissions into the atmosphere. ISRN Atmos. Sci. 2013, 2013, 1–27. [Google Scholar] [CrossRef]
- Williams, J.; Brune, W. A roadmap for OH reactivity research. Atmos. Environ. 2015, 106, 371–372. [Google Scholar] [CrossRef]
- Fuchs, H.; Novelli, A.; Rolletter, M.; Hofzumahaus, A.; Pfannerstill, E.Y.; Kessel, S.; Edtbauer, A.; Williams, J.; Michoud, V.; Dusanter, S.; et al. Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR. Atmos. Meas. Tech. 2017, 10, 4023–4053. [Google Scholar] [CrossRef]
- Yang, Y.; Shao, M.; Wang, X.; Nölscher, A.C.; Kessel, S.; Guenther, A.; Williams, J. Towards a quantitative understanding of total OH reactivity: A review. Atmos. Environ. 2016, 134, 147–161. [Google Scholar] [CrossRef]
- Karl, M.; Guenther, A.; Köble, R.; Seufert, G.; Leip, A.; Seufert, G. A new European plant-specific emission inventory of biogenic volatile organic compounds for use in atmospheric transport models. Biogeosciences Discuss. 2009, 5, 4993–5059. [Google Scholar] [CrossRef]
- Ren, X.; Brune, W.H.; Cantrell, C.A.; Edwards, G.D.; Shirley, T.; Metcalf, A.R.; Lesher, R.L. Hydroxyl and peroxy radical chemistry in a rural area of Central Pennsylvania: Observations and model comparisons. J. Atmos. Chem. 2005, 52, 231–257. [Google Scholar] [CrossRef]
- Lou, S.; Holland, F.; Rohrer, F.; Lu, K.; Bohn, B.; Brauers, T.; Chang, C.C.; Fuchs, H.; Häseler, R.; Kita, K.; et al. Atmospheric OH reactivities in the Pearl River Delta-China in summer 2006: Measurement and model results. Atmos. Chem. Phys. 2010, 10, 11243–11260. [Google Scholar] [CrossRef]
- Chameides, W.; Lindsay, R.W.; Richardson, J.; Kiang, C.S. The role of biogenic hydrocarbons in urban photochemical smog: Atlanta as a case study. Science 1988, 1473–1475. [Google Scholar] [CrossRef]
- Kim, S.; Guenther, A.; Karl, T.; Greenberg, J. Contributions of primary and secondary biogenic VOC tototal OH reactivity during the CABINEX (Community Atmosphere-Biosphere INteractions Experiments)-09 field campaign. Atmos. Chem. Phys. 2011, 11, 8613–8623. [Google Scholar] [CrossRef]
- Nölscher, A.C.; Bourtsoukidis, E.; Bonn, B.; Kesselmeier, J.; Lelieveld, J.; Williams, J. Seasonal measurements of total OH reactivity emission rates from Norway spruce in 2011. Biogeosciences 2013, 10, 4241–4257. [Google Scholar] [CrossRef]
- FAOSTAT. Available online: http://www.fao.org/faostat/en/#data/EL/visualize (accessed on 4 December 2020).
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef]
- Rodionova, M.V.; Poudyal, R.S.; Tiwari, I.; Voloshin, R.A.; Zharmukhamedov, S.K.; Nam, H.G.; Zayadan, B.K.; Bruce, B.D.; Hou, H.J.M.; Allakhverdiev, S.I. Biofuel production: Challenges and opportunities. Int. J. Hydrogen Energy 2017, 42, 8450–8461. [Google Scholar] [CrossRef]
- De Bouille, P.; Sotta, B.; Miginiac, E.; Merrien, A. Hormones and pod development in oilseed rape (brassica napus). Plant. Physiol. 1989, 90, 876–880. [Google Scholar] [CrossRef]
- McEwan, M.; Macfarlane Smith, W.H. Identification of volatile organic compounds emitted in the field by oilseed rape (Brassica napus ssp. oleifera) over the growing season. Clin. Exp. Allergy 1998, 28, 332–338. [Google Scholar] [CrossRef]
- Agreste, La Statistique, L’évaluation et la Prospective Agricole du Ministère de L’agriculture et de L’alimentation, Produits Agroalimentaires. Available online: https://agreste.agriculture.gouv.fr/agreste-web/download/publication/publie/GraFraChap12.1/Graf1941%20-%20COP.pdf (accessed on 4 December 2020).
- Müller, K.; Pelzing, M.; Gnauk, T.; Kappe, A.; Teichmann, U.; Spindler, G.; Haferkorn, S.; Jahn, Y.; Herrmann, H. Monoterpene emissions and carbonyl compound air concentrations during the blooming period of rape (Brassica napus). Chemosphere 2002, 49, 1247–1256. [Google Scholar] [CrossRef]
- Parratt, D.; Smith, W.M.; Thomson, G.; Cameron, L.A.; Butcher, R.D. Evidence that oilseed rape (brassica napus ssp. oleifera) causes respiratory illness in rural dwellers. Scottich Med. J. 1995, 40, 74–76. [Google Scholar] [CrossRef]
- Gomez, L.G.; Loubet, B.; Lafouge, F.; Ciuraru, R.; Buysse, P.; Durand, B.; Gueudet, J.C.; Fanucci, O.; Fortineau, A.; Zurfluh, O.; et al. Comparative study of biogenic volatile organic compounds fluxes by wheat, maize and rapeseed with dynamic chambers over a short period in northern France. Atmos. Environ. 2019, 214, 116855. [Google Scholar] [CrossRef]
- Vuolo, M.R.; Loubet, B.; Mascher, N.; Gueudet, J.C.; Durand, B.; Laville, P.; Zurfluh, O.; Ciuraru, R.; Stella, P.; Trebs, I. Nitrogen oxides and ozone fluxes from an oilseed-rape management cycle: The influence of cattle slurry application. Biogeosciences 2017, 14, 2225–2244. [Google Scholar] [CrossRef]
- Loubet, B.; Decuq, C.; Personne, E.; Massad, R.S.; Flechard, C.; Fanucci, O.; Mascher, N.; Gueudet, J.C.; Masson, S.; Durand, B.; et al. Investigating the stomatal, cuticular and soil ammonia fluxes over a growing tritical crop under high acidic loads. Biogeosciences 2012, 9, 1537–1552. [Google Scholar] [CrossRef]
- Laufs, S.; Cazaunau, M.; Stella, P.; Kurtenbach, R.; Cellier, P.; Mellouki, A.; Loubet, B.; Kleffmann, J. Diurnal fluxes of HONO above a crop rotation. Atmos. Chem. Phys. 2017, 17, 6907–6923. [Google Scholar] [CrossRef]
- Genard-Zielinski, A.C.; Boissard, C.; Fernandez, C.; Kalogridis, C.; Lathière, J.; Gros, V.; Bonnaire, N.; Ormeño, E. Variability of BVOC emissions from a Mediterranean mixed forest in southern France with a focus on Quercus pubescens. Atmos. Chem. Phys. 2015, 15, 431–446. [Google Scholar] [CrossRef]
- ICOS. Ecosystem Thematic Center, Documents. Available online: http://www.icos-etc.eu/icos/documents/ (accessed on 4 December 2020).
- Sinha, V.; Williams, J.; Crowley, J.N.; Lelieveld, J. The comparative reactivity method-A new tool to measure total OH Reactivity in ambient air. Atmos. Chem. Phys. 2008, 8, 2213–2227. [Google Scholar] [CrossRef]
- Atkinson, R. Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions. Chem. Rev. 1985, 85, 69–201. [Google Scholar] [CrossRef]
- Michoud, V.; Hansen, R.F.; Locoge, N.; Stevens, P.S.; Dusanter, S. Detailed characterizations of the new Mines Douai comparative reactivity method instrument via laboratory experiments and modeling. Atmos. Meas. Tech. 2015, 8, 3537–3553. [Google Scholar] [CrossRef]
- Praplan, A.P.; Pfannerstill, E.Y.; Williams, J.; Hellén, H. OH reactivity of the urban air in Helsinki, Finland, during winter. Atmos. Environ. 2017, 169, 150–161. [Google Scholar] [CrossRef]
- Zannoni, N.; Dusanter, S.; Gros, V.; Sarda Esteve, R.; Michoud, V.; Sinha, V.; Locoge, N.; Bonsang, B. Intercomparison of two comparative reactivity method instruments in the Mediterranean basin during summer 2013. Atmos. Meas. Tech. 2015, 8, 3851–3865. [Google Scholar] [CrossRef]
- Zannoni, N. OH Reactivity Measurements in the Mediterranean Region. Ph.D. Thesis, Paris-Saclay University, Paris, Ile-de-France, France, 2015. [Google Scholar]
- Abis, L.; Loubet, B.; Ciuraru, R.; Lafouge, F.; Dequiedt, S.; Houot, S.; Maron, P.A.; Bourgeteau-Sadet, S. Profiles of volatile organic compound emissions from soils amended with organic waste products. Sci. Total Environ. 2018, 636, 1333–1343. [Google Scholar] [CrossRef]
- Sulzer, P.; Hartungen, E.; Hanel, G.; Feil, S.; Winkler, K.; Mutschlechner, P.; Haidacher, S.; Schottkowsky, R.; Gunsch, D.; Seehauser, H.; et al. A proton transfer reaction-quadrupole interface time-of-flight mass spectrometer (PTR-QiTOF): High speed due to extreme sensitivity. Int. J. Mass Spectrom. 2014, 368, 1–5. [Google Scholar] [CrossRef]
- Pang, X. Biogenic volatile organic compound analyses by PTR-TOF-MS: Calibration, humidity effect and reduced electric field dependency. J. Environ. Sci. (China) 2015, 32, 196–206. [Google Scholar] [CrossRef]
- Tani, A.; Hayward, S.; Hewitt, C.N. Measurement of monoterpenes and related compounds by proton transfer reaction-mass spectrometry (PTR-MS). Int. J. Mass Spectrom. 2003, 223, 561–578. [Google Scholar] [CrossRef]
- Holzinger, R.; Acton, W.J.F.; Bloss, W.J.; Breitenlechner, M.; Crilley, L.R.; Dusanter, S.; Gonin, M.; Gros, V.; Keutsch, F.N.; Kiendler-Scharr, A.; et al. Validity and limitations of simple reaction kinetics to calculate concentrations of organic compounds from ion counts in PTR-MS. Atmos. Meas. Tech. Discuss. 2019, 1, 1–29. [Google Scholar] [CrossRef]
- Gros, V.; Gaimoz, C.; Herrmann, F.; Custer, T.; Williams, J.; Bonsang, B.; Sauvage, S.; Locoge, N.; d’Argouges, O.; Sarda-Estève, R.; et al. Volatile organic compounds sources in Paris in spring 2007. Part I: Qualitative analysis. Environ. Chem. 2011, 8, 74. [Google Scholar] [CrossRef]
- Assan, S.; Baudic, A.; Guemri, A.; Ciais, P.; Gros, V.; Vogel, F.R. Characterization of interferences to in situ observations of δ13 CH4 and C2H6 when using a cavity ring-down spectrometer at industrial sites. Atmos. Meas. Tech. 2017, 10, 2077–2091. [Google Scholar] [CrossRef]
- Warneke, C.; Veres, P.; Holloway, J.S.; Stutz, J.; Tsai, C.; Alvarez, S.; Rappenglueck, B.; Fehsenfeld, F.C.; Graus, M.; Gilman, J.B.; et al. Airborne formaldehyde measurements using PTR-MS: Calibration, humidity dependence, inter-comparison and initial results. Atmos. Meas. Tech. Discuss. 2011, 4, 4631–4665. [Google Scholar] [CrossRef]
- Atkinson, R.; Aschmann, S.M.; Arey, J. Rate constants for the gas-phase reactions of OH and NO3 radicals and O3 with sabinene and camphene at 296 ± 2 K. Atmos. Environ. Part. A, Gen. Top. 1990, 24, 2647–2654. [Google Scholar] [CrossRef]
- Atkinson, R.; Aschmann, S.M.; Pitts Jr, J.N. Rate constants for the gas-phase reactions of the OH radical with a series of monoterpenes at 294 ± 1 K. Int. J. Chem. Kinet. 1986, 18, 287–299. [Google Scholar] [CrossRef]
- Scala, A.; Allmann, S.; Mirabella, R.; Haring, M.A.; Schuurink, R.C. Green leaf volatiles: A plant’s multifunctional weapon against herbivores and pathogens. Int. J. Mol. Sci. 2013, 14, 17781–17811. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II–gas phase reactions of organic species. Atmos. Chem. Phys. 2006, 6, 3625–4055. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated kinetic and photochemical data for atmospheric chemistry: Part 1–gas phase reactions of Ox, HOx, NOx and SOx species. Atmos. Chem. Phys. Discuss. 2003, 3, 6179–6699. [Google Scholar] [CrossRef]
- Peeters, J.; Boullart, W.; Pultau, V.; Vandenberk, S.; Vereecken, L. Structure-activity relationship for the addition of OH to (poly)alkenes: Site-specific and total rate constants. J. Phys. Chem. A 2007, 111, 1618–1631. [Google Scholar] [CrossRef]
- Bignozzi, C.A.; Maldotti, A.; Chiorboli, C.; Bartocci, C.; Carassiti, V. Kinetics and mechanism of reactions between aromatic olefins and hydroxyl radicals. Int. J. Chem. Kinet. 1981, 13, 1235–1242. [Google Scholar] [CrossRef]
- Wilson, W.A.; Hamilton, H.R.; Kennington, B.; Evans, N.W.S.; Demore, W.B. Measurement and estimation of rate constants for the reactions of hydroxyl radical with several alkanes and cycloalkanes. J. Phys. Chem. A 2006, 110, 3593–3604. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Michael, J.V. Rate constants for OH with selected large alkanes: Shock-tube measurements and an improved group scheme. J. Phys. Chem. A 2009, 113, 5047–5060. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, D.; William, L.E. Environmental persistence of organic compounds estimated from structure-reactivity and linear free-energy relationships. Unsaturated aliphatics. Atmos. Environ. Part. A Gen. Top. 1992, 26, 1395–1405. [Google Scholar] [CrossRef]
- Atkinson, R.; Arey, J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: A review. Atmos. Environ. 2003, 37, 197–219. [Google Scholar] [CrossRef]
- Bsaibes, S.; Al Ajami, M.; Mermet, K.; Truong, F.; Batut, S.; Hecquet, C.; Dusanter, S.; Léornadis, T.; Sauvage, S.; Kammer, J.; et al. Variability of hydroxyl radical (OH) reactivity in the Landes maritime pine forest: Results from the LANDEX campaign 2017. Atmos. Chem. Phys. 2020, 20, 1277–1300. [Google Scholar] [CrossRef]
- Edwards, P.M.; Evans, M.J.; Furneaux, K.L.; Hopkins, J.; Ingham, T.; Jones, C.; Lee, J.D.; Lewis, A.C.; Moller, S.J.; Stone, D.; et al. OH reactivity in a South East Asian tropical rainforest during the oxidant and particle photochemical processes (OP3) project. Atmos. Chem. Phys. 2013, 13, 9497–9514. [Google Scholar] [CrossRef]
- Ingham, T.; Goddard, A.; Whalley, L.K.; Furneaux, K.L.; Edwards, P.M.; Seal, C.P.; Self, D.E.; Johnson, G.P.; Read, K.A.; Lee, J.D.; et al. A flow-tube based laser-induced fluorescence instrument to measure OH reactivity in the troposphere. Atmos. Meas. Tech. 2009, 2, 465–477. [Google Scholar] [CrossRef]
- Font, X.; Artola, A.; Sánchez, A. Detection, composition and treatment of volatile organic compounds from waste treatment plants. Sensors 2011, 11, 4043–4059. [Google Scholar] [CrossRef]
- König, G.; Brunda, M.; Puxbaum, H.; Hewitt, C.N.; Duckham, S.C.; Rudolph, J. Relative contribution of oxygenated hydrocarbons to the total biogenic VOC emissions of selected mid-European agricultural and natural plant species. Atmos. Environ. 1995, 29, 861–874. [Google Scholar] [CrossRef]
- Di Carlo, P.; Brune, W.H.; Martinez, M.; Harder, H.; Lesher, R.; Ren, X.; Thornberry, T.; Carroll, M.A.; Young, V.; Shepson, P.B.; et al. Missing OH Reactivity in a Forest: Evidence for Unknown Reactive Biogenic VOCs. Science 2004, 304, 722–725. [Google Scholar] [CrossRef]
- Guenther, A.B.; Zimmerman, P.R.; Harley, P.C.; Monson, R.K.; Fall, R. Isoprene and monoterpene emission rate variability: Model evaluations and sensitivity analyses. J. Geophys. Res. 1993, 98, 12609. [Google Scholar] [CrossRef]
- D’Anna, B.; Andresen; Gefen, Z.; Nielsen, C.J. Kinetic study of OH and NO3 radical reactions with 14 aliphatic aldehydes. Phys. Chem. Chem. Phys. 2001, 3, 3057–3063. [Google Scholar]
- Darnall, R.K.; Winer, A.M.; Lloyd, A.C.; Pitts, J.N. Relative rate constants for the reaction of OH radicals with selected C6 and C7 alkanes and alkenes at 305 ± 2 K. Chem. Phys. Lett. 1976, 44, 415–418. [Google Scholar] [CrossRef]
- Kwok, E.S.C.; Atkinson, R.; Arey, J. Kinetics of the gas-phase reactions of indan, indene, fluorene, and 9, 10-dihydroanthracene with OH radicals, NO3 radicals, and O3. Int. J. Chem. Kinet. 1997, 29, 299–308. [Google Scholar] [CrossRef]
- Chiorboll, C.A.; Bignozzi, A.; Maldotti, P.F.; Giardini, A.R. Rate constants for the gas-phase reactions of OH radicals with β-dimethylstyrene and acetone. Mechanism of β-dimethylstyrene NOx-air photooxidation. Int. J. Chem. Kinet. 1983, 15, 579–586. [Google Scholar] [CrossRef]
- Phousongphouang, P.T.; Arey, J. Rate constants for the gas-phase reactions of a series of alkylnaphthalenes with the OH radical. Environ. Sci. Technol. 2002, 36, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.; Tuazon, E.; Aschmann, S. Atmospheric chemistry of 2-Pentanone and 2-Heptanone. Environ. Sci. Technol. 2000, 34, 623–631. [Google Scholar] [CrossRef]
- Veromann, E.; Toome, M.; Kännaste, A.; Kaasik, R.; Copolovici, L.; Flink, J.; Kovács, G.; Narits, L.; Luik, A.; Niinemets, Ü. Effects of nitrogen fertilization on insect pests, their parasitoids, plant diseases and volatile organic compounds in Brassica napus. Crop. Prot. 2013, 43, 79–88. [Google Scholar] [CrossRef]
- Morrison, E.C.; Drewer, J.; Heal, M.R. A comparison of isoprene and monoterpene emission rates from the perennial bioenergy crops short-rotation coppice willow and Miscanthus and the annual arable crops wheat and oilseed rape. GCB Bioenergy 2016, 8, 211–225. [Google Scholar] [CrossRef]
- Kammer, J.; Décuq, C.; Baisnée, D.; Ciuraru, R.; Lafouge, F.; Buysse, P.; Bsaibes, S.; Henderson, B.; Cristescu, S.M.; Benabdallah, R.; et al. Characterization of the particle and gaseous pollutants emissions from a French dairy and sheep farm. Sci. Total Environ. 2019, 712, 135598. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Measurement | Time Resolution |
|---|---|---|
| CRM | Total OH Reactivity (ROH) | 10 min |
| PTRQi-ToFMS | VOCs Formaldehyde, Methanol, Acetaldehyde, Butanol, Acetone, Acetic acid, Isoprene, MethylEthylKetone (MEK), Hexadiene/ GLV, Styrene, Monoterpenes | 0.1 s |
| GC-FID | C2–C6 HCs Alkanes: Ethane, Propane, i-butane, n-butane, i-pentane, n-pentane, 2-methylpentane, n-hexane Alkenes: Ethene, Propene, Trans-2-butene, 1-butene, i-butene, Cis-2-butene, 1,3-butadiene, Trans-2-pentene, 1-pentene Alkynes: Acetylene | 30 min |
| NO-NO2-NOx chemiluminescence analyzer | NO, NO2 | 1 min |
| G2201-i Picarro | CH4 | 1 min |
| 48i-TL, thermo-Environnement | CO | 1 min |
| Measurements | Protonated Theoretical Mass | Chemical Compound | Name | Rate Constant (× 10−12 cm3 molecule−1 s−1) | Ref. |
|---|---|---|---|---|---|
| PTRQi-ToFMS protonated masses | m31.018 | (CH2O)H+ | Formaldehyde | 8.5 | [50] |
| m33.033 | (CH3OH)H+ | Methanol | 0.9 | [50] | |
| m45.033 | (CH3CHO)H+ | Acetaldehyde | 15 | [50] | |
| m57.069 | (C4H8)H+ | Butanol fragment | 8.5 | [50] | |
| m59.049 | (C3H6O)H+ | Acetone | 0.18 | [50] | |
| m61.029 | (C2H4O2)H+ | Acetic Acid | 0.74 | [50] | |
| m69.070 | (C5H8)H+ | Isoprene | 100 | [50] | |
| m73.064 | (C4H8O)H+ | Butanone/ MEK | 1.2 | [50] | |
| m81.068 | (C6H8)H+ | MTs fragment | 165 | [51] | |
| m83.084 | (C6H10)H+ | Hexadiene/C6 GLV | 69.1 | [52] | |
| m105.066 | (C8H8)H+ | Styrene | 53 | [53] | |
| m137.129 | (C10H16)H+ | Monoterpenes (MTs) | 165 | [51] | |
| GC-FID | C2H6 | Ethane | 0.24 | [50] | |
| C2H4 | Ethene | 7.9 | [50] | ||
| C3H8 | Propane | 1.1 | [50] | ||
| C3H6 | Propene | 29 | [50] | ||
| C4H10 | i-butane | 2.35 | [51] | ||
| C4H10 | n-butane | 2.35 | [51] | ||
| C2H2 | Acetylene | 0.75 | [51] | ||
| (E)-2-C4H8 | Trans-2-butene | 63.7 | [34] | ||
| 1- C4H8 | 1-butene | 31.5 | [34] | ||
| I- C4H8 | i-butene | 51.5 | [34] | ||
| (Z)-2-C4H8 | Cis-2-butene | 56 | [34] | ||
| iso-C5H12 | i-pentane | 3.65 | [54] | ||
| n-C5H12 | n-pentane | 3.92 | [55] | ||
| CH2=CH-CH=CH2 | 1,3-butadiene | 58 | [56] | ||
| 2- (E)- C5H10 | Trans-2-pentene | 57.1 | [56] | ||
| 1-C5H10 | 1-pentene | 35 | [56] | ||
| (CH3)2CH(CH2)2CH3 | 2-Me-pentane | 5.2 | [57] | ||
| C6H14 | n-hexane | 5.38 | [55] | ||
| Other compounds | CO | Carbon monoxide | 0.228 | [51] | |
| CH4 | Methane | 0.0064 | [51] | ||
| NO | Nitrogen oxide | 9.7 | [51] | ||
| NO2 | Nitrogen dioxide | 11.3 | [51] |
| ROH and ROHmissing | Daytime Avg ± STD | Night-time Avg ± STD |
|---|---|---|
| cROH NOx (s−1) | 3.6 ± 2.2 | 3.8 ± 0.9 |
| cROH HCs C2- C6 (s−1) | 0.4 ± 0.2 | 0.5 ± 0.2 |
| cROH CO (s−1) | 0.8 ± 0.1 | 0.9 ± 0.1 |
| cROH CH4 (s−1) | 0.3 ± 0.01 | 0.3 ± 0.07 |
| cROH VOCambient (s−1) | 0.8 ± 0.7 | 1.5 ± 1.0 |
| cROH VOCrapeseed (s−1) | 1.9 ± 1.7 | <LOD ± 0.7 |
| Mean cROHchamber (s−1) | 7.8 ± 2.6 | 7.0 ± 1.5 |
| Mean mROHchamber (s−1) | 14.4 ± 7.0 | 8.0 ± 2.6 |
| Mean missing ROH (s−1) | 6.6 ± 5.0 | 1.0 ± 1.7 |
| Mean relative missing ROH (%) | 46 | 13 |
| Protonated Theoretical Mass | Chemical Formula | Chemical Compound | Rate Constant (× 10−12 cm3 Molecule−1 s−1) | Max Mixing Ratio (ppbv) | Max Calculated ROH (s−1) | Reference |
|---|---|---|---|---|---|---|
| 57.033 | C3H4O | Propenal | 19.6 | 0.23 | 0.11 | [34] |
| 85.064 | C5H8O | Ethylvinylketone | 36 a | 0.06 | 0.06 | [56] |
| 85.1 | C6H12 | Hexene | 37 a | 0.18 | 0.17 | [56] |
| 87.078 | C5H10O | 2-Methylbutanal b | 32.8 c | 0.06 | 0.05 | [65] |
| 97.098 | C7H12 | Methylcyclohexene | 96.1 d | 0.3 | 0.66 | [66] |
| 109.1 | C8H12 | 4-Ethenylcyclohexene | 63.9 a | 0.23 | 0.36 | [56] |
| 119.082 | C9H10 | Indan | 19.1 e | 0.02 | 0.01 | [67] |
| 133.098 | C10H12 | Benzene, (2-methyl−1-propenyl)- | 33 | 0.01 | 0.01 | [68] |
| 157.105 | C12H12 | Dimethylnaphtalene | 74.9 f | 0.03 | 0.06 | [69] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bsaibes, S.; Gros, V.; Truong, F.; Boissard, C.; Baisnée, D.; Sarda-Esteve, R.; Zannoni, N.; Lafouge, F.; Ciuraru, R.; Buysse, P.; et al. Characterization of Total OH Reactivity in a Rapeseed Field: Results from the COV3ER Experiment in April 2017. Atmosphere 2020, 11, 261. https://doi.org/10.3390/atmos11030261
Bsaibes S, Gros V, Truong F, Boissard C, Baisnée D, Sarda-Esteve R, Zannoni N, Lafouge F, Ciuraru R, Buysse P, et al. Characterization of Total OH Reactivity in a Rapeseed Field: Results from the COV3ER Experiment in April 2017. Atmosphere. 2020; 11(3):261. https://doi.org/10.3390/atmos11030261
Chicago/Turabian StyleBsaibes, Sandy, Valérie Gros, François Truong, Christophe Boissard, Dominique Baisnée, Roland Sarda-Esteve, Nora Zannoni, Florence Lafouge, Raluca Ciuraru, Pauline Buysse, and et al. 2020. "Characterization of Total OH Reactivity in a Rapeseed Field: Results from the COV3ER Experiment in April 2017" Atmosphere 11, no. 3: 261. https://doi.org/10.3390/atmos11030261
APA StyleBsaibes, S., Gros, V., Truong, F., Boissard, C., Baisnée, D., Sarda-Esteve, R., Zannoni, N., Lafouge, F., Ciuraru, R., Buysse, P., Kammer, J., Gomez, L. G., & Loubet, B. (2020). Characterization of Total OH Reactivity in a Rapeseed Field: Results from the COV3ER Experiment in April 2017. Atmosphere, 11(3), 261. https://doi.org/10.3390/atmos11030261