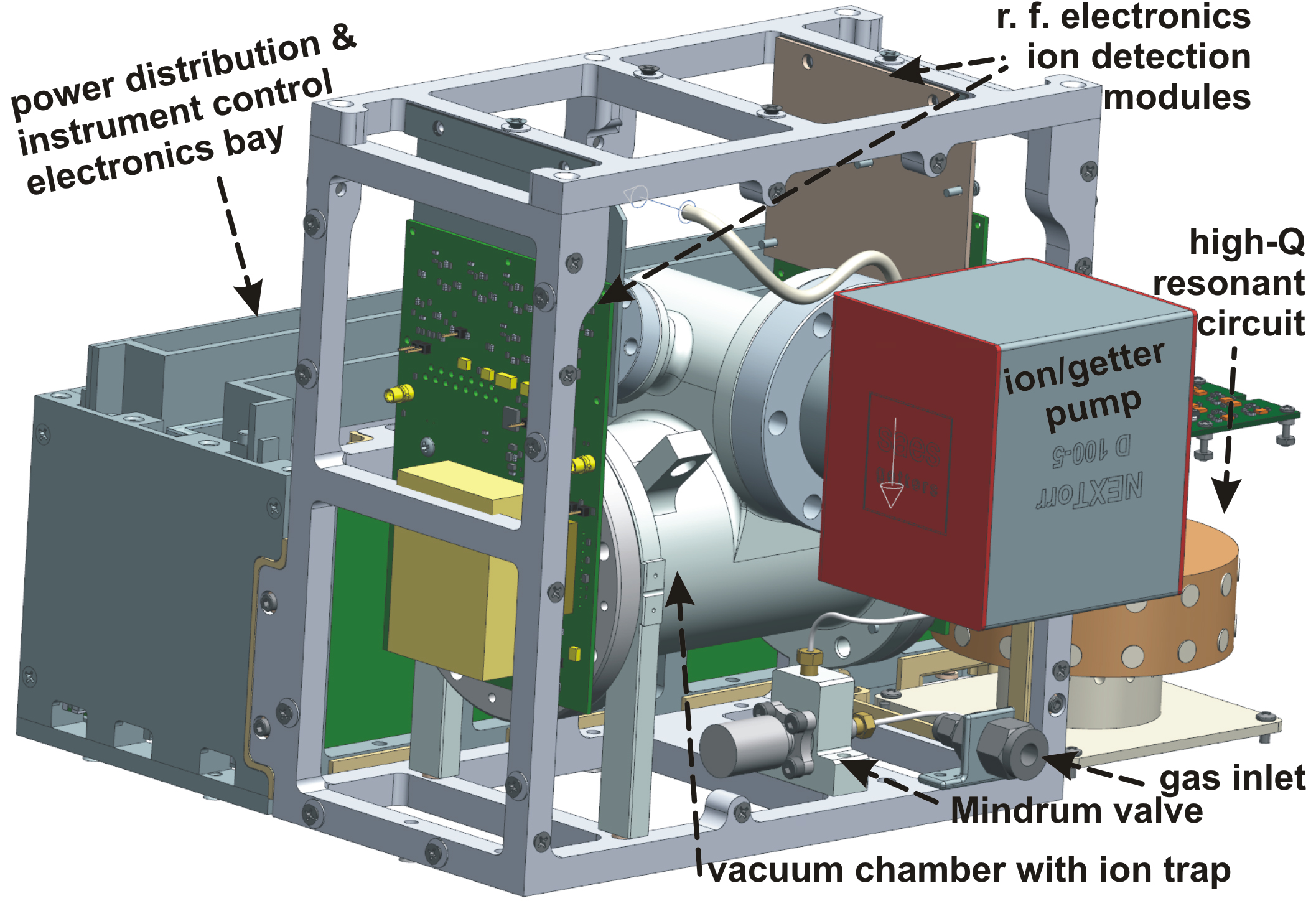
The time-dependent physical properties of atmospheric sample serve as an input to the Sample Acquisition and Handling (SA&H) module. The SA&H module deals with sample thermalization and simulates the non-evaporable getter pump using variable sorption efficiency of chemically-active species. The output of the SA&H module is the composition of the sample volume that depends on already adsorbed chemically-active species. The sample volume is then released into the QITMS-S module. This module simulates the dynamics of gas flow into the vacuum chamber, controls the getter pump adsorbtion dynamics, provides the ion generation, ion confinement, and ion analysis and finally performs the analysis of isotopic ratios.
3.2. Compositional Model of Venus Atmosphere
As illustrated in
Figure 2, second important input to the SA&H module is the composition of the Venus atmosphere. The time-dependent partial pressures of molecular species entering the QIT-MS-I are derived from the published data on the composition of Venus atmosphere and the simulated probe trajectory, see Equation (1). Note that the total pressure at the inlet of the instrument is a sum of static and dynamic pressures. The static pressure at 110 km is about 0.096 Pa. This value is derived from number densities of Kransopolsky [
26,
27] and mean VIRA [
25] daytime (LST = 12 h) and nighttime (LST = 0 h) temperature data for 100–150 km altitudes. The dynamic pressure is about 4500 times larger than the static pressure at the 110 km altitude, which gives 437.3 Pa as suggested by the average mean VIRA [
25] mass density, re-normalized atmospheric composition, and probe velocity.
In the current model, an increased temperature and density of gas sample in the bow shock are considered insufficient for thermal decomposition of CO
2 [
28,
29], noble gas partition [
30], or charge transfer reactions [
31,
32] between noble gas atoms and CO
2 molecules. In general, thermal decomposition of CO
2 in the bow shock may include CO, O
2 and C produced in three reactions [
28],
where the values of equilibrium constants K
1, K
2, and K
3 were calculated at each temperature
(up to 6000 K) using relation
and NIST-JANAF [
29] free energy of formation data.
The coefficients
,
, and
are shown in
Table 1, and when compared to
Table 2 of Ref. [
28], they yield no precipitation of carbon and up to 6% larger values for the mole fractions of CO and O
2 produced by dissociation of 1 mole of CO
2 along the phase boundary curves. Small difference is due to an extended temperature range (100–6000 K) and revised data on the free energy of formation [
33]. Along the phase boundary curve, mole fraction of CO and O
2 are in range of few parts in 1 × 10
13, and can be neglected as source of uncertainty for native CO and O
2 abundances. Thermal ionization of noble gases in the bow shock is also neglected as an unlikely contribution to isotope fractionation due to the cross-shock potential [
34], but will be revisited in a future study.
The approximate chemical composition of the Venus atmosphere at 110 km altitude is shown in
Table 2 and
Table 3. The variation of all chemically-active trace species below 112 km altitude is adopted from the photo-chemical model of Krasnopolsky [
26,
27]. Altitude dependent isotopic abundances of noble gases are due to Hoffman et al. [
35,
36], Wieler [
37], Fegley [
38], and Donahue et al. [
39] or where unavailable, except
78Kr, substituted with terrestrial values due to De Laeter et al. [
40] and Wieser et al. [
41]. Decision to leave out
78Kr comes from large uncertainties in atmospheric models containing isobars C
6H
6 and CS
2 and warrants a separate study. The unknown thermal production rates in the hypervelocity high-temperature (6 × 10
4 K) bow shock as well as unknown gettering rates for these molecules contribute to unknown interference levels at
m/
q 78 which will be addressed in a future study. Here we assume that all chemically active species except noble gases will be removed from the gas sample by getter pumps. The calculated molecular isotope abundances are based on the algorithm of Hugentobler & Loliger [
42] and are readily obtained for molecules of arbitrary chemical complexity.
Blending different data sources to infer relative abundances of chemical species requires their re-normalization. Namely, with every 0.5 km drop in altitude starting from 112 km down to 110 km, mixing ratios of noble gases and trace species were balanced by the binary mixture of 96.5% CO
2 and 3.5% N
2 and re-normalized to unity. Using the atmospheric temperature of 175 K, atmospheric mass density of 7.56 × 10
−6 kg/m
3, and corresponding mixing ratios, we compute the intake partial pressures for each chemical species as given in
Table 2 and
Table 3. These intake pressures will set the pV-flow through the sampling inlet of the SA&H module.
3.3. Model of Sample Acquisition and Handling
The next simulation phase in
Figure 2 deals with the SA&H module. The pathway of Venus atmosphere sample through the SA&H module is illustrated in
Figure 2. The pathway consists of the sampling inlet followed by the puncture valve and line to the Mindrum microvalve (M
1). After M
1, the sample first enters the thermalizer and then the holding tank, which is isolated from QIT-MS-S by M
2 microvalve. In this simulation phase, we replace with zero-length tube the sample line leading to the puncture valve, the puncture valve itself, and the line leading from the puncture valve to and including the M
1 valve. In addition, the sample pathway between M
1 and the holding tank and thermalizer are in our model simplified by a single stainless steel tube (SST) with length
L = 10 cm and inner diameter
d = 1 mm (of volume
Vs) held at room temperature (300 K). The holding tank is made of the 304 stainless steel and has volume
Vh = 1ℓ and mass of 15 g. The M
2 valve is connected via zero-length tubes to the sampling volume on one side and to the vacuum chamber on the other side. The realistic CO
2 flow rate through M
1/M
2 valves, as a function of the head pressure and the pumping speed of getter pumps, will be reported in a future study.
Prior to the sampling, the vacuum chamber remains isolated from the holding tank by the M
2 valve, whereas the holding tank is isolated from the sampling inlet by the M
1 valve. At this point, both the holding tank and the vacuum chamber contain only the residual gas, which is assumed here to be sustained at the background pressure of
= 3.2 × 10
−8 Pa. This estimate is based on the following composition of the background pressure [
43]: H
2 (83.2%), H
2O (5.9%), N
2 (0.8%), CO (4.4%), CO
2 (5.7%). The exact compositions of the residual background pressure need to be measured, monitored, and corrected for as part of each static pressure measurement. Possible mitigations include coating of vacuum chamber’s interior surfaces by getter materials. Each of these molecular species (with mass
m) will contribute its own partial pressure
to the total background pressure
. The vacuum of the system is maintained by CapaciTorr D100 getter pump [
44] directly attached to the holding tank as well as ion pump / getter combination (NEXTorr D100-5) connected to the vacuum chamber (when M
2 is open). Estimated amount of gas (1Torr = 133.322 Pa) that getters need to adsorb during 2 years of background vacuum maintenance is α
H2 = 1.2 Torrℓ and α
CO2 = 0.08 Torrℓ, which is well below the single run getter capacity [
45].
In the following we will describe the calculation leading to the sample volume composition, which is an output from the SA&H module. The atmosphere sampling begins by opening the
valve for 2 s, see
Table 4. The duration of sampling depends on several, mutually interconnected requirements: (a) the anticipated inlet pressure at the lowest point in trajectory; (b) the model used to describe conductivity of the sampling inlet tube [
46]; (c) the volume of the holding tank; (d) the maximum single run capacity of the getter pump used to remove CO
2 and N
2 from the holding tank; (e) the total pressure of all noble gas species in the sample upon its release into the vacuum chamber for analysis; and most importantly, (f) the scientific requirements on precision for noble gas isotopic ratios.
For a given inlet pressure, the flow of atmospheric sample is established through the sampling inlet tube. Dimensions of the inlet tube can be chosen in accordance to the total amount of gas that can be admitted into the holding tank without saturating the getter pump with CO2 and N2. Otherwise, the getter pump will saturate and pumping speeds for these dominant active species will remain at about 10% of their maximum values. The choice of the flow model is crucial in establishing the amount of gas that has been admitted into the holding tank via the sampling inlet. If inadequate model of molecular flow is used, even when M1 remains open for sufficiently long time, the partial pressure of any noble gas isotope inside the holding tank will increase asymptotically only to the value at the sampling inlet, and no further enrichment is possible. The enrichment of the noble gases in the holding tank is achievable only if the dissolved flow model is used, such that the back flow of trace isotopes out of the holding tank is inhibited.
In the dissolved flow model, see
Section 3.3.2, we assume all minor species are dissolved in binary CO
2/N
2 gas mixture, and according to Equation (3), will be carried through the sampling tube within the directional flow. As long as the partial pressure of CO
2 inside the holding tank remains lower than its inlet value, no back flow of dissolved noble gas isotopes is possible. The CO
2 pressure difference
that is required to maintain the directional flow is sustained by the CapaciTorr D100 getter pump [
44]. Depending on the amount of admitted sample, all noble gas isotopes can be enriched at various levels by adjusting the duration of sampling. For example, sampling through
L = 10 cm long inlet tube for 2 s can achieve 11.6 factor of enrichment for
128Xe inside the holding tank as compared to the inlet value, but it will require tube with diameter
d = 6.5 mm and getter pump capable of adsorbing 37 Torr ℓ of mostly CO
2 and N
2. For tube with the diameter
d = 4 mm the enrichment factor is 1.7 for
128Xe compared to the inlet value and getter will be required to adsorb 5.4 Torr ℓ. If we instead use a tube with the diameter
d = 3 mm then the depletion factor for
128Xe is 0.54 compared to the inlet, whereas the getter must adsorb 1.8 Torr ℓ.
The depletion of partial pressures inside the holding tank is a consequence of a short sampling time for a given inlet tube conductance. Even with the depleted noble gas sample, as compared to its inlet composition, the dissolved flow model yields 1.77 times higher 128Xe partial pressure in the holding tank compared to the molecular flow model. Therefore, for a given form factor of the sampling inlet, we can control the amount of admitted gas into the holding tank by adjusting the time for which M1 valve remains open. Depending on the amount of admitted gas, the isotope content in the sample compared to the inlet, can be conveniently enriched or depleted. However, the admitted amount of sample must always be below the single-run adsorption capacity for the Capacitor getter. It is important to note that use of dissolved flow dynamics instead of molecular flow approximation is required for assessing the accuracy.
3.3.1. Micro Valve State Logic
Mindrum valves are operated according to the sequence given in
Table 4. The zero time is defined as the time when atmospheric probe is at 200 km altitude. The sampling of Venus atmosphere begins at 65.9 s when the probe’s altitude is 110 km, see Equation (1).
At the beginning of the survey, see the first row in
Table 4, the M
1 valve is open during 2 s admitting the sample of atmosphere into the holding tank, whereas the M
2 valve remains closed protecting the QIT-MS-S from the inlet pressure surge. After 2 s, see the second row in
Table 4, both the M
1 and M
2 valves are closed for 9.6 s during which gettering of CO
2/N
2 and other chemically-active species is performed. When scrubbing is finished at
= 77.5 s, the sample containing mostly noble gases is admitted into the vacuum chamber for mass spectrometry analysis by opening the M
2 valve, see the third row in
Table 4. Upon the completion of the analysis, the ion pump is activated and both the vacuum chamber and the holding tank are pumped out and prepared for the next flyby sampling.
3.3.2. Computation of Dissolved pV-flow
We simulate the
pV-flow
of gas particles (with molecular mass
) through narrow tubes of diameter
d and length
L using the closed form result of Fryer [
47]:
which is applicable to wide range of flow regimes, from molecular to viscous. The molecular mass
and temperature
T determine the value for the mean thermal component of gas velocity. The inlet pressure
pin governs the time-dependent directional component of velocity in tubular flow.
First term in Equation (3) accounts for the Poiseuille flow corrected for the proportion of molecules with mean free path larger than the tube diameter. Central term describes slip flow in which molecules that interact with tube walls are assumed to possess no average flow momentum. Last term in Equation (3) accounts for molecular diffusion near tube walls. The pressure gradient in Equation (3) implicitly includes the difference between the inlet and the outlet partial pressures, because directional flow will stop when these two pressures are equal. Outlet pressure is always assumed to be downstream from the inlet. The inlet pressure will change with the flyby time during sample admission into the holding tank, mainly due to hypervelocity changes in the atmosphere composition in the bow shock (dependent on probe velocity) whereas the outlet pressure will change due to the active pumping of the holding tank.
Inlet pressure
in Equation (3) is scaled by the characteristic pressure
:
The
is pressure at which molecular mean free path
~
(for a given molecular mass
m and temperature
T) equals the tube diameter
d, and is dominated by CO
2 gas. The collisional diameter
in Equation (4) is dependent on molecular mass
m, gas temperature, and gas pressure. We estimate the value for
using the gas viscosity
and the approximate relation
[
47], where
and
are mass density and mean molecular thermal velocity, respectively. The thermal and physical properties of CO
2 gas are well known [
48,
49,
50,
51,
52] and viscosity data can be conveniently tabulated for wide range of temperatures and pressures relevant to Venus’ atmosphere. Viscosity data for CO
2 gas are then used to estimate free mean path
, collisional molecular diameter
, and characteristic pressure
pc.
Although not considered in this work, a similar approach could be applied to all minor species listed in
Table 2 and
Table 3 whenever their viscosity data are known. However, detailed viscosity studies exist only for a few chemical species of interest to the present study. For example, Hanley [
53] reports the viscosity of dilute Ar, Kr, and Xe gases for temperatures from 2000 K down to about one-half the critical temperature. Similarly, for N
2 and O
2 viscosity, Cole & Wakeham [
54] report values in the zero-density limit for 110–2100 K temperature range. Viscosity for binary gas mixtures can be approximated using results of Brokaw [
55] and Davidson [
56], and in general can yield the corrections to the CO
2 diameter
for binary mixtures with nitrogen. For simplicity we neglect contribution of N
2 viscosity in CO
2/N
2 binary mixture because N
2 enters the mixture at 3.5%. Instead, when passing through the sampling inlet shown in
Figure 1, all minor species are regarded as fully dissolved in the dominant CO
2 matrix. By maintaining the sampling inlet flow at room temperature, the characteristic pressure
pc in Equation (4) is due to CO
2 and will change only with the tube diameter
d. For the fixed dimensions of the sampling tube, the
pV-flow
in Equation (3) will predominantly change with the inlet pressure
pin and the type of chemical species
m being transferred.
3.3.3. Computation of Pumping Speeds
The mass-dependent
pV-flow
through the SST of sampling inlet is driven by the time-dependent pressure difference between the head of the sampling inlet and the holding tank. Thus, the time-dependent
pV-flow is calculated with help of Equation (3), (4), such that the M
1 valve remains open during 2 s of sampling and admits 23 × 10
−3 Torrℓ of atmosphere into the holding tank, see
Figure 3. At the same time, downstream from the holding tank, the Mindrum valve M
2 remains closed, see
Table 4.
The holding tank is continuously pumped by the CapaciTorr D100 getter pump. We assume, without loss of generality, that the holding tank temperature
T = 300 K is unchanged by admiting hot/fast sample. This assumption is supported by the following consideration. The holding tank has the specific heat capacity of 0.5 Jg
−1 K
−1 [
57] and during 2 s of sampling at ~10 km/s it will accommodate about 7.4 × 10
17 molecules. Admitted molecules may exchange at most 3.2 J of its kinetic energy in collisions with holding tank walls, and consequently may raise the temperature of the system by at most 0.4 K.
The CapaciTorr D100 pumping speeds
as a function of already adsorbed material
have been previously measured by Hogan & Malyshev [
45] but only for a few common getterable gases (N
2, H
2, CO, CO
2, CH
4). Pumping speed for O
2 (
m = 32) can be found in the NEXTorr D100-5 user manual. In general, hydrogen and its isotopes are sorbed reversibly; CO, CO
2, O
2, and N
2 are chemisorbed irreversibly; H
2O and hydrocarbons are sorbed in a combination of slow reversible and irreversible processes, whereas inert gases are not sorbed at all and are enriched relative to the residual gas. Details about simulated pumping speeds of chemically-active species as a function of already adsorbed material will be published elsewhere.
3.4. Composition of Sample Volume
The composition of sample volume is the output from the simulated SA&H module, see
Figure 2, and consists of an inventory of all molecular species present in the sample as a function of time. The time-dependent number of molecules
of mass
inside the holding tank of volume
is obtained as numerical solution to the set of coupled first order differential equations,
where the molecular mass
takes values for isotopes of all chemical species found in
Table 2 and
Table 3. Coupling is governed by the instantaneous values of adsorption
, which represents amount of already sorbed material due to all species. The sampling inlet, holding tank, and vacuum chamber (with the volume of
= 0.371ℓ) are all maintained at room temperature,
= 300 K.
At the start of atmospheric measurements, see
Table 4, the initial background partial pressures
are assumed to be zero except for the previously discussed residual gas molecules. The solution of Equation (5) for the carbon dioxide
= CO
2 in the form of the time-dependent partial pressure
is further detailed in
Figure 3. At the instant when the M
2 valve is closed, the total amount of adsorbed gas is
≈ 23 ×10
−3 Torr ℓ, which will degrade the pumping speed for CO
2 to 88% of its unsaturated value; similar degradation in pumping speed is seen across other getterable chemical species, such are for example N
2 (85%) and S
8(92%). During the static measurement of noble gases, ion pump is turned off and the pumping speed for all noble gases is assumed to be zero.
Figure 3 illustrates that partial pressures of the most abundant noble gas isotopes will never exceed the 2.7 × 10
−4 Pa limit. If needed, this limit can be lowered most efficiently by reducing the inner diameter
d of the sampling inlet tube. As illustrated in
Figure 3, after opening the M
2 valve (
= 77.5 s), the sample will expand into the vacuum chamber and noble gas partial pressures will reach equilibrium levels that are
≈ 0.73 times the previous value.
3.5. Model of the QIT-MS Sensor
Modeling the performance of the QIT-MS sensor is the next phase in the simulation flow chart given in
Figure 2. The composition of the sample volume is previously obtained in the SA&H module, and is used here as an input to the simulation of the QIT-MS sensor capabilities. The Capacitor D100 getter pump attached to the holding tank and the NEXTorr D100-5 getter pump attached to the vacuum chamber efficiently adsorb any residual chemically-active species except noble gases.
Shortly after the M
2 is open, residual partial pressures of chemically-active species in both the holding tank and the vacuum chamber will assume a new equilibrium levels listed in
Table 5. The new background pressure will be the volume-weighted average of background pressures in the holding tank and in the vacuum chamber prior to the opening of the M
2 valve. For simplicity we will assume that the new background pressure remains unchanged as indicated in
Figure 3 for CO
2 and N
2, and reported in
Table 5 for other background species. At the same time noble gas isotopes in the sample volume will remain enriched at relative levels established previously in the holding tank as listed in
Table 6. The total pressure of all noble gas isotopes is equilibrated at 1.49 × 10
−4 Pa.
The least abundant isotopes
3He,
124Xe, and
126Xe contribute to the sample volume in amounts that are five orders of magnitude smaller than the
40Ar and
36Ar contributions. Accurate detection of trace amounts of isotopes in the presence of other dominant isotopes in a short period of time represents a challenge, as it requires a sufficient counting of statistics of rare events, namely, in the nominal operation of the QIT-MS sensor all ions are indiscriminately created, stored, and recorded 20 times per second [
15]. However, as previously discussed in
Section 2.2, the QIT-MS sensor operates with peak performance given that the following two conditions are fulfilled: (a) the space charge effects due to ion-ion interactions are negligible, and (b) ion collisions with neutral molecules are rare. We determined empirically [
15] that both conditions are satisfied when the total number of ions within the trap does not exceed 5000 at any given moment and the pressure does not exceed 2.7 × 10
−4 Pa limit.
Therefore, in the nominal operation, the repetition rate of 20 duty cycles per second amounts to at most 100,000 ions being analyzed each second. Note that the duration of 50 ms for the single cycle has been chosen to provide mass resolution of 800 at 140 u, which will fully separate individual isotope peaks in xenon [
15]. The mass resolution scales with the square root of analysis time. For example, if the duration of the trapping phase is kept at 6.12 ms, see
Figure 1c, but the duration of a duty cycle is reduced from 50 ms to 25 ms, the count rate will double and the mass resolution will be 52% lower. Lower mass resolution causes neighboring isotope peaks to overlap and counting integrated counts under the single peak becomes unreliable metrics for accurate determination of isotopic ratios [
15].
For each individual cycle we can shorten or prolong the duration of the ionization phase on demand, effectively adjusting the number of stored ions to below 5000. It is evident from
Table 6 that the
36Ar and
40Ar isotopes will contribute 85.8% of all created ions. On the other hand, the most abundant krypton isotope
84Kr will be created only 0.08% of the time, and thus, will contribute 4 ± 2 ions in each 50 ms duty cycle. To remedy this problem, we make use of resonant ejection technique [
16], such that during the ionization phase we simultaneously destabilize trajectories of select
m/
q ions.
This is done by adding two additional low-amplitude phase-inverted RF drives to the QIT-MS-S. For trapping conditions used in this study, the frequencies for removing 36Ar+ and 40Ar+ ions are 108.2 kHz and 96.9 kHz, respectively. The resonant ejection method is used in 19 out of 20 duty cycles to suppress 36Ar+ and 40Ar+ ions by factor of 100. We note that doubly-charged 36Ar2+ and 40Ar2+ ions will not be affected. The last duty cycle out of 20 duty cycles is carried out without the resonant ejection, but with much shorter ionization phase where only the most abundant isotopes are created and trapped. In this hybrid operation mode, we effectively improve the dynamic range of the instrument.
The starting point for calculation of the composition of the detected ion cloud is the neutral molecular/atomic composition represented with individual partial pressures
, see
Table 6. Using the trap sensitivity
s and the probability π
m for confinement and detection of
m/
q ions, we can calculate the number of ions being detected for each individual measurement cycle. Distribution of ionization rates of the sample species was obtained by using the recommended absolute total cross sections for electron-impact ionization of the noble gases [
58]. For example, by using the electron beam with 70 eV energy, He
2+ and Ne
2+ will not be created, whereas the Ar
1+/Ar
2+, Kr
1+/Kr
2+, and Xe
1+/Xe
2+ ion number ratios are estimated to be 18.4, 87.3, and 12.1 respectively. During 6.12 ms of the ion trapping phase, all created ions in the first 5 ms were confined using the RF potential with constant amplitude of 96.1 V. In the analysis phase, the RF amplitude was linearly ramped to 1049 V in 43.88 ms. For each ion we register its position, velocity, and time of detection. Initial ions are randomly generated during the first 5 ms of the trapping phase and uniformly distributed inside the cylinder (4 mm long axially and 1 mm wide radially, see inset in
Figure 1a) placed in the center of the QIT-MS sensor. In the present study the 4 mm axial spread of the initial ion cloud is chosen to benchmark the effect of overlap among neighboring isotopes in the mass spectrum. Initial thermal velocities of created ions are randomly drawn from the Maxwell-Boltzmann distribution at the temperature of 300 K.
Immediately after their creation, ions are propagated through the RF quadrupole electric field using 1 ns time steps. Ion velocities and positions are updated by modified Verlet algorithm [
16]. Propagation of ions is stopped if the duty cycle has ended, a collision with electrode surfaces has been recorded, or ion has been detected. At the end of every duty cycle, detected ions are added to the final ion cloud and post-processed to yield a measured mass spectrum. By comparing the number of initial ions of given m/q and the corresponding number of detected ions, we calculate the detection probability π
m.
Table 7 summarizes the average number of ions being detected in 1 s of probe flyby time or equivalently in 20 subsequent measurement cycles. The total counting rate for noble gases is
s×
= 99263 cps, which means that every second on average we are randomly sampling from the Poisson distribution 99263 ions within standard uncertainty of 315 ions. To avoid oversampling, the pool of 300 million ions has been pre-generated by the Computational Ion Trap Analyzer [
16] (CITA) such that end-cap electrodes are grounded and the central ring electrode is driven by the 867.791 kHz RF potential. Sample pool of ions when
36Ar
+ and
40Ar
+ ions are resonantly depleted is also created by CITA program by driving the end-caps by an auxiliary phase-inverted RF potential of 0.6 V amplitude and with mixed dual frequency (108.2 kHz and 96.9 kHz).
Response of the QIT-MS instrument to neutral gas sample is accumulated in 1 s intervals by twenty random samplings of approximately 5000 ions from the final ion cloud using Poisson probability density function. As an example,
Table 8 illustrates the distribution of detected ions in twenty subsequent duty cycles. Sampled mass spectra can be co-added in time intervals of up to 40 min in duration; then we extract ion detection times and mass-to-charge (
m/
q) ratios and perform a linear regression analysis by computing the slope and the intercept of the regression line through data. This procedure establishes the linear correspondence between the ion’s detection time and its
m/
q value. By counting how many ions of certain
m/
q value will contribute to the given
m/
q bin width, we generate histogram plots representing a mass spectrum, see
Figure 1. For example, 2724 counts for
38Ar
+ that are detected in the first duty cycle of
Table 8 will be all distributed in 15 bins each 1 × 10
−2 u wide. In the nominal QIT-MS data acquisition setting, after 40 min of operation we will accumulate a count matrix with 40 × 60 × 20 = 48,000 rows (duty cycles) such that each row is representing a full mass spectrum with 16,000 mass bins. Each row in the data matrix is statistically independent from any other row, which is a direct consequence of Monte-Carlo simulation approach when creating the ion cloud for each duty cycle. Namely, initial ion positions, thermal velocities, and creation times will always be different between duty cycles, and detected mass spectra will be statistically different.
We note that collisions among ions and neutral molecules are neglected as rare events [
15], but can be included in propagation of ions to study the spread and broadening of line shapes of each mass peak.
Figure 4 illustrates the part of synthetic mass spectrum accumulated in 40 min of static measurements of noble gas isotopes. Data matrix has been histogrammed in 1 × 10
−2 u wide bins. For comparison, the typical mass spectra provided by previously flown mass-hopping spectrometer [
59] are of 1u resolution.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}