Epigenetic Control of Pancreatic Regeneration in Diabetes

{kind=link}
Abstract
:1. Introduction
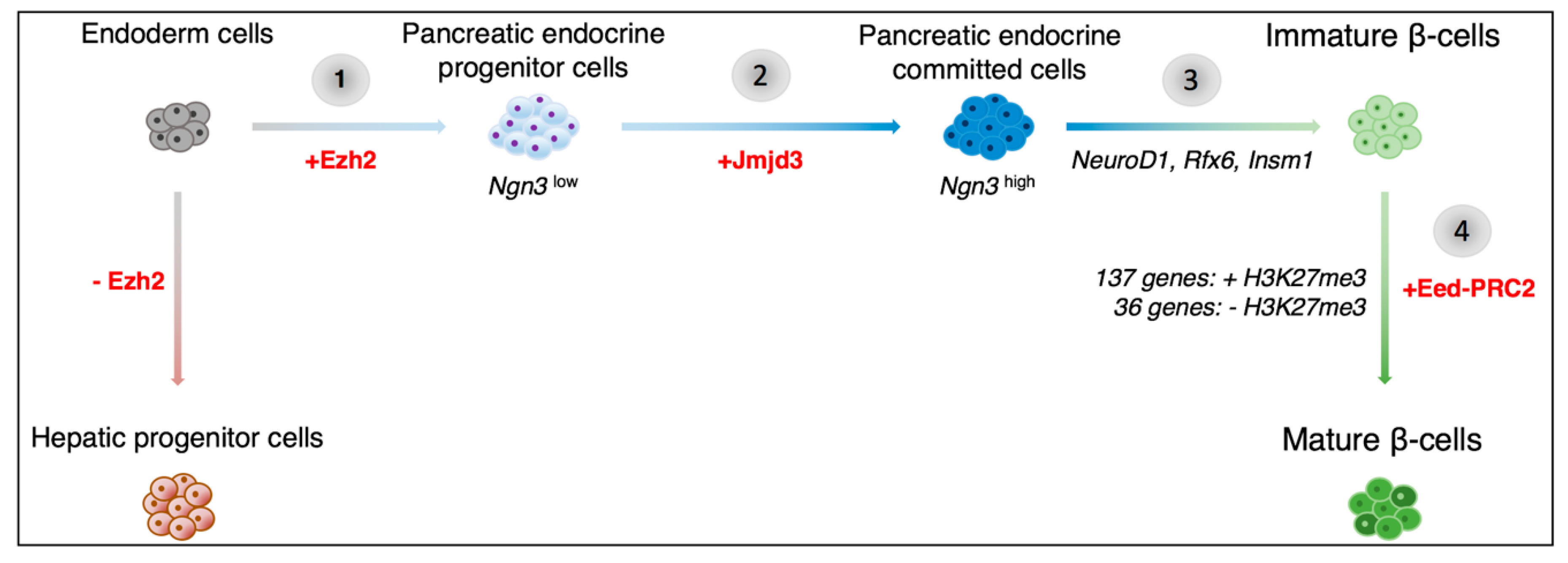
2. Advances in Our Understanding of the Epigenetics of Pancreas Development
3. The Epigenetic State in Type 2 Diabetes and Obesity
4. Epigenetics and Pancreatic Regeneration
5. The Influence of Culture Substrate on Epigenetic Regulation of Differentiation
6. Conclusions
Funding
Conflicts of Interest
References
- Gittes, G.K. Developmental biology of the pancreas: A comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Bonner-Weir, S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyk, A.C.; Giacomini, K.N.; McKeon, C.; Shuldiner, A.R.; Florez, J.C. Metformin pharmacogenomics: Current status and future directions. Diabetes 2014, 63, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, L.; Fleming, G.A.; Petrie, J.R.; Holl, R.W.; Bergenstal, R.M.; Peters, A.L. Insulin pump risks and benefits: A clinical appraisal of pump safety standards, adverse event reporting, and research needs. Diabetes Care 2015, 38, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Heller, S.R.; Miller, M.A. Treatment of type 2 diabetes mellitus in the older adult: A review. Endocr. Pract. 2014, 20, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Zhou, Y.; Opara, E.C.; Soker, S. Combinations of activin A or nicotinamide with the pancreatic transcription factor PDX1 support differentiation of human amnion epithelial cells toward a pancreatic lineage. Cell. Reprogram. 2017, 19, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Sambathkumar, R.; Akkerman, R.; Dastidar, S.; Roelandt, P.; Kumar, M.; Bajaj, M.; Rosa, A.R.M.; Helsen, N.; Vanslembrouck, V.; Kalo, E.; et al. Generation of hepatocyte- and endocrine pancreatic-like cells from human induced endodermal progenitor cells. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and Subsequently β Cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, K.; Pfeifer, A.; Courtney, M.; Ben-Othman, N.; Gjernes, E.; Vieira, A.; Druelle, N.; Avolio, F.; Ravassard, P.; Leuckx, G.; et al. Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell 2013, 26, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Zhou, Y.; Ganguly, A.; Opara, E.C.; Soker, S. The combined effect of PDX1, epidermal growth factor and poly-l-ornithine on human amnion epithelial cells’ differentiation. BMC Dev. Biol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Carrano, A.C.; Sander, M. A systems view of epigenetic networks regulating pancreas development and β-cell function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quilichini, E.; Haumaitre, C. Implication of epigenetics in pancreas development and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 883–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The role of epigenetics in type 1 diabetes. Curr. Diab. Rep. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48. [Google Scholar] [CrossRef] [PubMed]
- Hiragami-Hamada, K.; Fischle, W. RNAs—Physical and functional modulators of chromatin reader proteins. Biochim. Biophys. Acta 2014, 1839, 737–742. [Google Scholar] [CrossRef] [PubMed]
- James, L.I.; Frye, S.V. Targeting chromatin readers. Clin. Pharmacol. Ther. 2013, 93, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, U.; Jonsson, J.; Edlund, H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996, 122, 1409–1416. [Google Scholar] [PubMed]
- Collombat, P.; Mansouri, A.; Hecksher-Sørensen, J.; Serup, P.; Krull, P.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pineda, B.; Chowdhury, K.; Torres, M.; Oliver, G.; Gruss, P. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature 1997, 386, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-R.; Cole, P.A.; Meyers, D.J.; Kormish, J.; Dent, S.; Zaret, K.S. Chromatin “pre-pattern” and histone modifiers in a fate choice for liver and pancreas. Science 2011, 332, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.R.; Li, L.C.; Donahue, G.; Ying, L.; Zhang, Y.W.; Gadue, P.; Zaret, K.S. Dynamics of genomic H3K27me3 domains and role of EZH2 during pancreatic endocrine specification. EMBO J. 2014, 33, 2157–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Arensbergen, J.; García-Hurtado, J.; Moran, I.; Maestro, M.A.; Xu, X.; Van de Casteele, M.; Skoudy, A.L.; Palassini, M.; Heimberg, H.; Ferrer, J. Derepression of polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 2010, 20, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Qiu, W.L.; Yang, L.; Li, L.C.; Zhang, Y.W.; Xu, C.R. Dynamics of chromatin marks and the role of JMJD3 during pancreatic endocrine cell fate commitment. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of β cells. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-L.; Zhang, Y.-W.; Feng, Y.; Li, L.-C.; Yang, L.; Xu, C.-R. Deciphering Pancreatic Islet β Cell and α Cell Maturation Pathways and Characteristic Features at the Single-Cell Level. Cell Metab. 2017, 25, 1194–1205.e4. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human α cell and β cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Neiman, D.; Moss, J.; Hecht, M.; Maggenheim, J.; Piyanzin, S.; Shapiro, A.M.J.; de Koning, E.J.P.; Razin, A.; Cedar, H.; Shemer, R.; et al. Islet cells share promoter hypomethylation independently of expression, but exhibit cell-type-specific methylation in enhancers. Proc. Natl. Acad. Sci. USA 2017, 114, 13525–13530. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Heyne, S.; Dror, E.; Casas, E.; Leonhardt, L.; Boenke, T.; Yang, C.H.; Sagar; Arrigoni, L.; Dalgaard, K.; et al. The polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 2018, 27, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Li, C.; Zhang, J.; Schug, J.; Avrahami, R.; Rao, S.; Sadler, M.B.; Burger, L.; Schübeler, D.; Glaser, B.; et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015, 22, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Voight, B.F.; Teslocvich, T.M.; Ferreira, T.; Segré, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooner, J.S.; Saleheen, D.; Sim, X.; Sehmi, J.; Zhang, W.; Frossard, P.; Been, L.F.; Chia, K.S.; Dimas, A.S.; Hassanali, N.; et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 2011, 43, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellado-Gil, J.M.; Fuente-Martín, E.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; López-Noriega, L.; Martín-Montalvo, A.; Gómez, I.G.H.; Ceballos-Chávez, M.; Gómez-Jaramillo, L.; Campos-Caro, A.; et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet β cell functional maturity. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Grapin-Botton, A. The importance of REST for development and function of β cells. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic β-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Kahn, C.R. The Molecular Mechanism of Insulin Action. Annu. Rev. Med. 1985, 36, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisbis, S.; Bailbe, D.; Tormo, M.A.; Picarel-Blanchot, F.; Derouet, M.; Simon, J.; Portha, B. Insulin resistance in the GK rat: Decreased receptor number but normal kinase activity in liver. Am. J. Physiol. Endocrinol. Metab. 1993, 265, E807–E813. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.T.; Zhou, Y.; Evertts, A.; Xu, S.; Griffin, M.J.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, Y.; Feng, Y.; Long, D.; Ma, K.; Wang, X.; Zhao, M.; Lu, L.; Lu, Q. Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.N.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Huang, G.; Wang, Z.; Luo, S.; Zheng, P.; Zhou, Z. Epigenetic regulation of Toll-like receptors and its roles in type 1 diabetes. J. Mol. Med. 2018, 96, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Zurawek, M.; Dzikiewicz-Krawczyk, A.; Izykowska, K.; Ziolkowska-Suchanek, I.; Skowronska, B.; Czainska, M.; Podralska, M.; Fichna, P.; Przybylski, G.; Fichna, M.; et al. miR-487a-3p upregulated in type 1 diabetes targets CTLA4 and FOXO3. Diabetes Res. Clin. Pract. 2018, 142, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell. Mol. Immunol. 2018, 15, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontcuberta-PiSunyer, M.; Cervantes, S.; Miguel, E.; Mora-Castilla, S.; Laurent, L.C.; Raya, A.; Gomis, R.; Gasa, R. Modulation of the endocrine transcriptional program by targeting histone modifiers of the H3K27me3 mark. Biochim. Biophys. Acta 2018, 1861, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gu, X.; Su, I.H.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieria, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic α-Cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Georgia, S.; Tschen, S.I.; Fan, G.; Bushan, A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell 2011, 20, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting adult pancreatic islet α cells into β cells by targeting both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Leong, K.W. Biomaterials approach to expand and direct differentiation of stem cells. Mol. Ther. 2007, 15, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Pennarossa, G.; Santoro, R.; Manzoni, E.F.M.; Pesce, M.; Gandolfi, F.; Brevini, T.A.L. Epigenetic erasing and pancreatic differentiation of dermal fibroblasts into insulin-producing cells are boosted by the use of low-stiffness substrate. Stem Cell Rev. 2018, 14, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Kortbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; Oaty, B.W.; Senior, P.A.; Bigam, D.; Alfadhli, E.; Kneteman, N.M.; Lakey, J.R.; Shapiro, A.M. Five-year follow-up after clinical islet transplantation. Diabetes 2005, 54, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mack, D.L.; Williams, J.K.; Mirmalek-Sani, S.H.; Moorefield, E.; Chun, S.Y.; Wang, J.; Lorenzetti, D.; Furth, M.; Atala, A.; et al. Genetic modification of primate amniotic fluid-derived stem cells produces pancreatic progenitor cells in vitro. Cells Tissues Organs 2013, 197, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Cito, M.; Pellegrini, S.; Piemonti, L.; Sordi, V. The potential and challenges of alternative sources of β cells for the cure of type 1 diabetes. Endocr. Connect. 2018, 7, R114–R125. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balaji, S.; Napolitano, T.; Silvano, S.; Friano, M.E.; Garrido-Utrilla, A.; Atlija, J.; Collombat, P. Epigenetic Control of Pancreatic Regeneration in Diabetes. Genes 2018, 9, 448. https://doi.org/10.3390/genes9090448
Balaji S, Napolitano T, Silvano S, Friano ME, Garrido-Utrilla A, Atlija J, Collombat P. Epigenetic Control of Pancreatic Regeneration in Diabetes. Genes. 2018; 9(9):448. https://doi.org/10.3390/genes9090448
Chicago/Turabian StyleBalaji, Shruti, Tiziana Napolitano, Serena Silvano, Marika Elsa Friano, Anna Garrido-Utrilla, Josipa Atlija, and Patrick Collombat. 2018. "Epigenetic Control of Pancreatic Regeneration in Diabetes" Genes 9, no. 9: 448. https://doi.org/10.3390/genes9090448
APA StyleBalaji, S., Napolitano, T., Silvano, S., Friano, M. E., Garrido-Utrilla, A., Atlija, J., & Collombat, P. (2018). Epigenetic Control of Pancreatic Regeneration in Diabetes. Genes, 9(9), 448. https://doi.org/10.3390/genes9090448