A Computational Method for Classifying Different Human Tissues with Quantitatively Tissue-Specific Expressed Genes

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Feature Ranking and Selection
2.3. Classification Algorithm
2.4. Measurements
3. Results
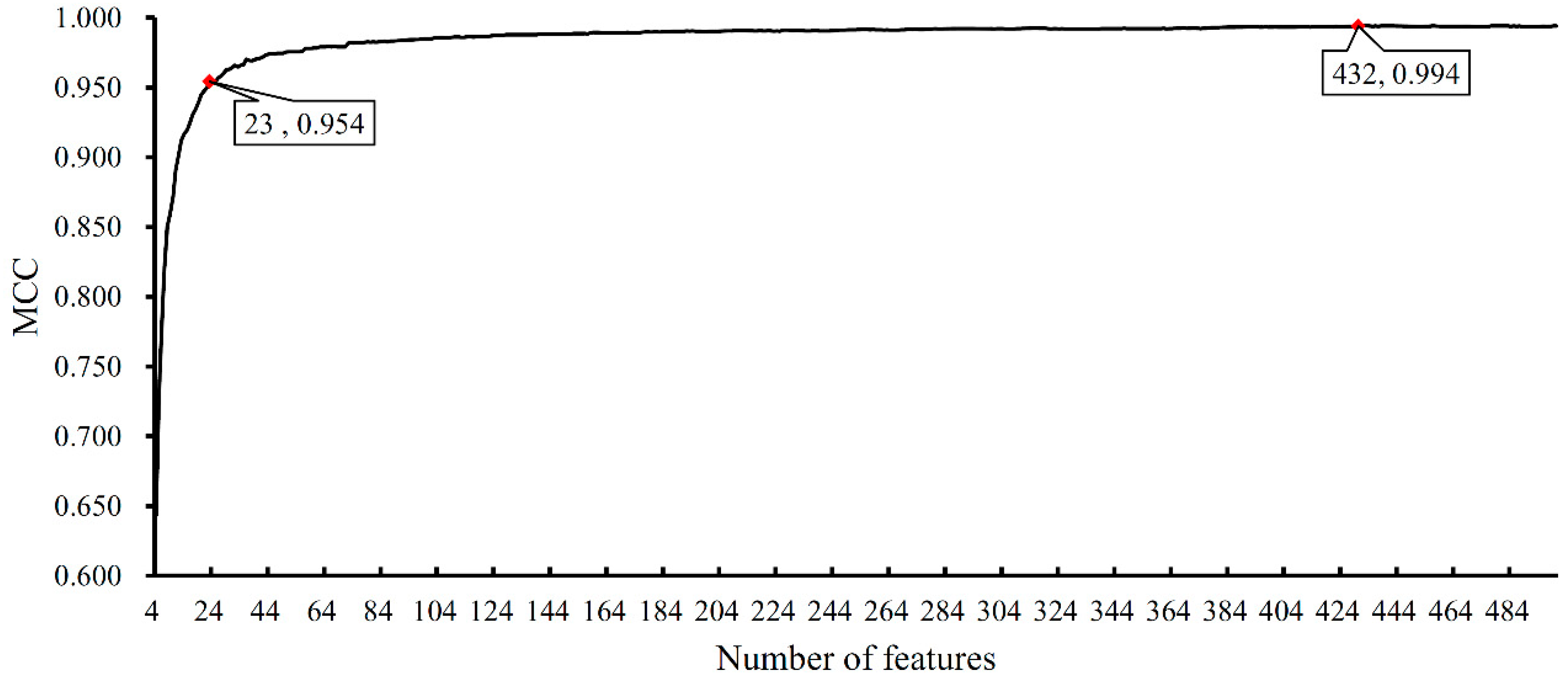
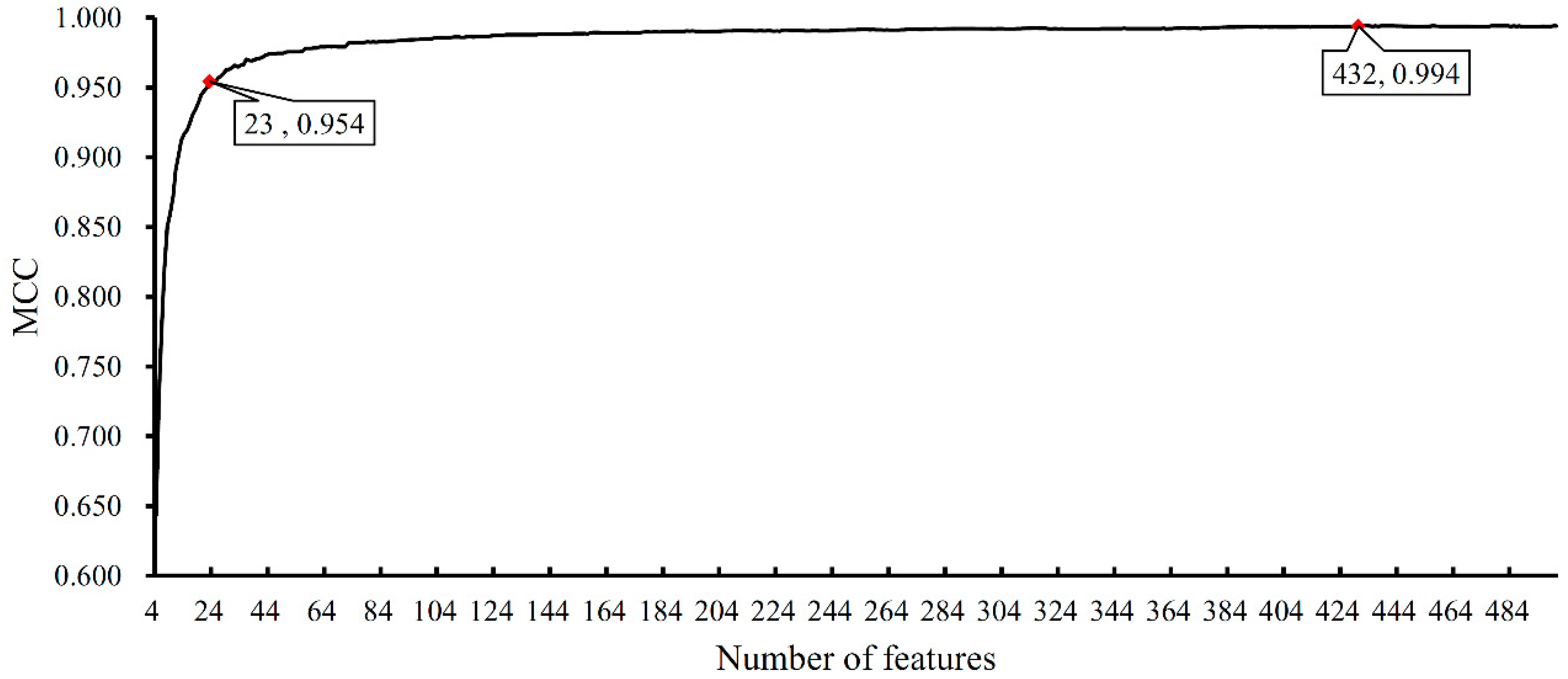
3.1. Results of Feature Ranking
3.2. Results of Feature Selection
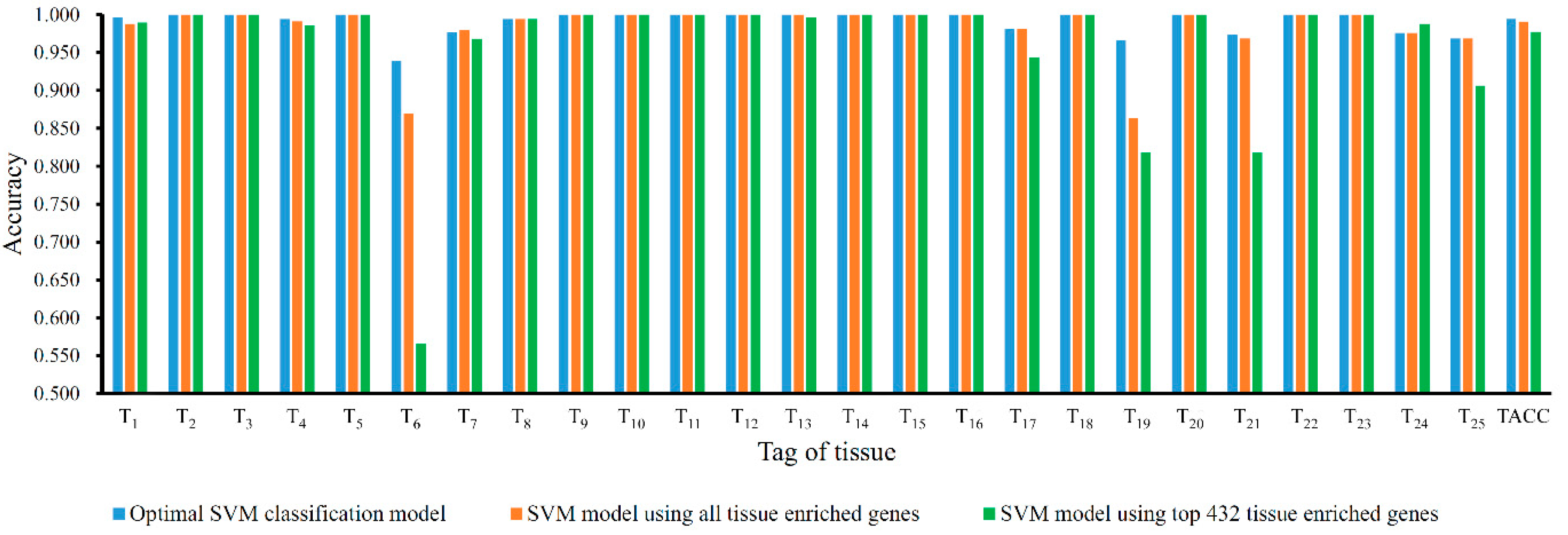
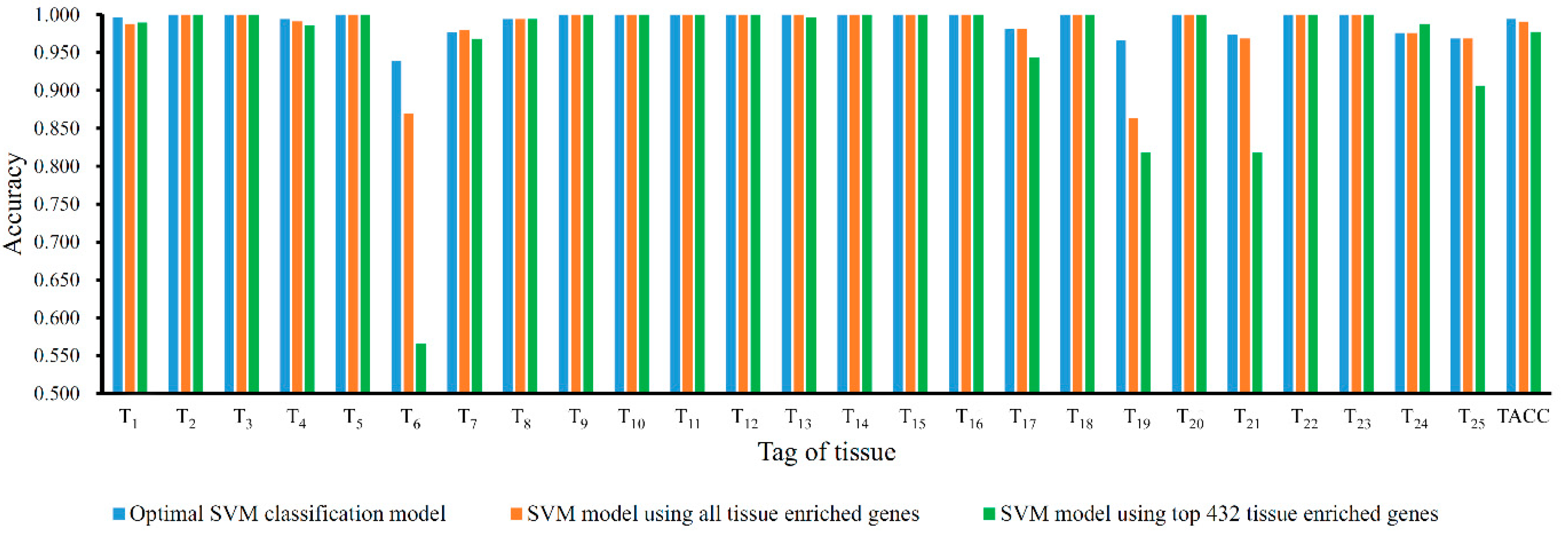
3.3. Comparison of SVM Model with Tissue Enriched Genes
3.4. Performance of the Optimal SVM Classification Model on Test Dataset
3.5. Comparison of SVM Model with t-Test Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Singh, S.R. Stem cell niche in tissue homeostasis, aging and cancer. Curr. Med. Chem. 2012, 19, 5965–5974. [Google Scholar] [CrossRef] [PubMed]
- Lipscombe, D.; Andrade, A. Calcium channel cavα1 splice isoforms—Tissue specificity and drug action. Curr. Mol. Pharmacol. 2015, 8, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. MCP 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., III; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Krupp, M.; Marquardt, J.U.; Sahin, U.; Galle, P.R.; Castle, J.; Teufel, A. RNA-seq atlas—A reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics 2012, 28, 1184–1185. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium; Human genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Leisch, F.; Hornik, K. The support vector machine under test. Neurocomputing 2003, 55, 169–186. [Google Scholar] [CrossRef]
- Corinna, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Cai, Y.D.; Feng, K.Y.; Zhao, G.J. Prediction of protein cleavage site with feature selection by random forest. PLoS ONE 2012, 7, e45854. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.-H.; Lu, G.; Huang, T.; Cai, Y.-D. Analysis of cancer-related lncRNAs using gene ontology and kegg pathways. Artif. Intell. Med. 2017, 76, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; He, J.; Lu, L. Predicting sumoylation site by feature selection method. J. Biomol. Struct. Dyn. 2011, 28, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.-H.; Huang, G.; Pan, X.; Wang, S.; Huang, T.; Cai, Y.-D. Discriminating cirRNAs from other lncRNAs using a hierarchical extreme learning machine (H-ELM) algorithm with feature selection. Mol. Genet. Genom. 2018, 293, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, S.; Cai, Y.D.; Zhang, Q. Analysis and prediction of nitrated tyrosine sites with mRMR method and support vector machine algorithm. Curr. Bioinform. 2017, 13, 3–13. [Google Scholar]
- Liu, L.; Chen, L.; Zhang, Y.H.; Wei, L.; Cheng, S.; Kong, X.; Zheng, M.; Huang, T.; Cai, Y.D. Analysis and prediction of drug-drug interaction by minimum redundancy maximum relevance and incremental feature selection. J. Biomol. Struct. Dyn. 2017, 35, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.H.; Huang, T.; Cai, Y.D. Gene expression profiling gut microbiota in different races of humans. Sci. Rep. 2016, 6, 23075. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.; Chen, L. A feature and algorithm selection method for improving the prediction of protein structural classes. Comb. Chem. High Throughput Screen. 2017, 20, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.H.; Zheng, M.; Huang, T.; Cai, Y.D. Identification of compound-protein interactions through the analysis of gene ontology, kegg enrichment for proteins and molecular fragments of compounds. Mol. Genet. Genom. 2016, 291, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Y.-H.; Huang, G.; Chen, L.; Cai, Y.-D. Analysis and prediction of myristoylation sites using the mRMR method, the ifs method and an extreme learning machine algorithm. Comb. Chem. High Throughput Screen. 2017, 20, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Wei, L.; Xu, X.; Huang, T.; Cai, Y.-D. Prediction of nitrated tyrosine residues in protein sequences by extreme learning machine and feature selection methods. Comb. Chem. High Throughput Screen. 2018, 21, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Zheng, L.L.; Hu, L.L.; Feng, K.Y.; Huang, G.; Chen, L. Prediction of linear B-ceel epitopes with mRMR feature selection and analysis. Curr. Bioinform. 2016, 11, 22–31. [Google Scholar] [CrossRef]
- Chen, L.; Pan, X.; Hu, X.; Zhang, Y.-H.; Wang, S.; Huang, T.; Cai, Y.-D. Gene expression differences among different MSI statuses in colorectal cancer. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Platt, J. Sequential Minimal Optimizaton: A Fast Algorithm for Training Support Vector Machines; Technical Report MSR-TR-98-14; Microsoft Res: Redmon, WA, USA, 1998. [Google Scholar]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Lawrence Erlbaum Associates Ltd.: Mahwah, NJ, USA, 1995; pp. 1137–1145. [Google Scholar]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Li, J.; Xing, Z.-H.; Yang, J.; Huang, T.; Cai, Y.-D. Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access 2017, 5, 26582–26590. [Google Scholar] [CrossRef]
- Wang, D.; Li, J.-R.; Zhang, Y.-H.; Chen, L.; Huang, T.; Cai, Y.-D. Identification of differentially expressed genes between original breast cancer and xenograft using machine learning algorithms. Genes 2018, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Zhang, Y.-H.; Zheng, M.-Y.; Zhu, L.; Kong, X.; Huang, T. Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 2017, 12, 526–534. [Google Scholar] [CrossRef]
- Chen, L.; Zeng, W.M.; Cai, Y.D.; Feng, K.Y.; Chou, K.C. Predicting anatomical therapeutic chemical (ATC) classification of drugs by integrating chemical-chemical interactions and similarities. PLoS ONE 2012, 7, e35254. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Gorodkin, J. Comparing two K-category assignments by a K-category correlation coefficient. Comput. Biol. Chem. 2004, 28, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- eGTEx Project; Stranger, B.E.; Brigham, L.E.; Hasz, R.; Hunter, M.; Johns, C.; Johnson, M.; Kopen, G.; Leinweber, W.F.; Lonsdale, J.T.; et al. Enhancing gtex by bridging the gaps between genotype, gene expression, and disease. Nat. Genet. 2017, 49, 1664. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Fullgrabe, A.; Fuentes, A.M.; George, N.; et al. Expression atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W. The role of atypical protein kinase C in CSF-1-dependent ERK activation and proliferation in myeloid progenitors and macrophages. PLoS ONE 2011, 6, e25580. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.H.; Xu, F.; Zhang, Q.A.; Wu, Z.Y.; Zhang, X.J.; Xu, J.H.; Luo, Y.; Guan, M. Oncogenic mutations in extramammary Paget’s disease and their clinical relevance. Int. J. Cancer 2013, 132, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lu, Z.Y.; Jia, J.Y.; Zheng, Z.F.; Lin, S. Changes in microRNAs associated with podocytic adhesion damage under mechanical stress. J. Renin-Angiotensin Aldosterone Syst. 2013, 14, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Pinatel, E.M.; Orso, F.; Penna, E.; Cimino, D.; Elia, A.R.; Circosta, P.; Dentelli, P.; Brizzi, M.F.; Provero, P.; Taverna, D. miR-223 is a coordinator of breast cancer progression as revealed by bioinformatics predictions. PLoS ONE 2014, 9, e84859. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, G.C.; Treadway, M.B.; Petrone, A.B.; Tennant, C.S.; Lucke-Wold, N.; Chantler, P.D.; Barr, T.L. Peripheral blood AKAP7 expression as an early marker for lymphocyte-mediated post-stroke blood brain barrier disruption. Sci. Rep. 2017, 7, 1172. [Google Scholar] [CrossRef] [PubMed]
- Van der Vaart, B.; Franker, M.A.M.; Kuijpers, M.; Hua, S.S.; Bouchet, B.P.; Jiang, K.; Grigoriev, I.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins SLAIN1/2 and ch-TOG promote axonal development. J. Neurosci. 2012, 32, 14722–14729. [Google Scholar] [CrossRef] [PubMed]
- Suchy-Dicey, A.; Heckbert, S.R.; Smith, N.L.; McKnight, B.; Rotter, J.I.; Chen, Y.I.; Psaty, B.M.; Enquobahrie, D.A. Gene expression in thiazide diuretic or statin users in relation to incident type 2 diabetes. Int. J. Mol. Epidemiol. Genet. 2014, 5, 22–30. [Google Scholar] [PubMed]
- Cowell, J.K.; Lo, K.C.; Luce, J.; Hawthorn, L. Interpreting aCGH-defined karyotypic changes in gliomas using copy number status, loss of heterozygosity and allelic ratios. Exp. Mol. Pathol. 2010, 88, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Ye, Z.; Gu, Y.; Tian, B.; Wu, B.; Li, J. Genomic analysis of drug resistant pancreatic cancer cell line by combining long non-coding RNA and mRNA expression profling. Int. J. Clin. Exp. Pathol. 2015, 8, 38–52. [Google Scholar] [PubMed]
- Gao, Y.F.; Zhu, T.; Mao, C.X.; Liu, Z.X.; Wang, Z.B.; Mao, X.Y.; Li, L.; Yi, J.Y.; Zhou, H.H.; Liu, Z.Q. PPIC, EMP3 and CHI3L1 are novel prognostic markers for high grade glioma. Int. J. Mol. Sci. 2016, 17, 1808. [Google Scholar] [CrossRef] [PubMed]
- Romero-Saavedra, F.; Laverde, D.; Wobser, D.; Michaux, C.; Budin-Verneuil, A.; Bernay, B.; Benachour, A.; Hartke, A.; Huebner, J. Identification of peptidoglycan-associated proteins as vaccine candidates for enterococcal infections. PLoS ONE 2014, 9, e111880. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Ben-Arie, N. A novel role for the choroid plexus in BMP-mediated inhibition of differentiation of cerebellar neural progenitors. Mech. Dev. 2006, 123, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ohtori, S.; Yamamoto, T.; Ino, H.; Hanaoka, E.; Shinbo, J.; Ozaki, T.; Takada, N.; Nakamura, Y.; Chiba, T.; Nakagawara, A.; et al. Differential screening-selected gene aberrative in neuroblastoma protein modulates inflammatory pain in the spinal dorsal horn. Neuroscience 2002, 110, 579–586. [Google Scholar] [CrossRef]
- Yi, C.H.; Zheng, T.Z.; Leaderer, D.; Hoffman, A.; Zhu, Y. Cancer-related transcriptional targets of the circadian gene NPAS2 identified by genome-wide ChIP-on-chip analysis. Cancer Lett. 2009, 284, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siggs, O.M.; Popkin, D.L.; Krebs, P.; Li, X.H.; Tang, M.; Zhan, X.M.; Zeng, M.; Lin, P.; Xia, Y.; Oldstone, M.B.A.; et al. Mutation of the er retention receptor kdelr1 leads to cell-intrinsic lymphopenia and a failure to control chronic viral infection. Proc. Natl. Acad. Sci. USA 2015, 112, E5706–E5714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, T.; Valle, D. Reduced PLP2 expression increases ER-stress-induced neuronal apoptosis and risk for adverse neurological outcomes after hypoxia ischemia injury. Hum. Mol. Genet. 2015, 24, 7221–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Miao, M.H.; Ji, X.Q.; Xue, J.; Shao, X.J. miR-664 negatively regulates PLP2 and promotes cell proliferation and invasion in T-cell acute lymphoblastic leukaemia. Biochem. Biophys. Res. Commun. 2015, 459, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, N.J.; Chapoval, S.P.; Smith, E.P.; Skupsky, J.; Scott, D.W.; Keegan, A.D. STAT6 controls the number of regulatory T cells in vivo, thereby regulating allergic lung inflammation. J. Immunol. 2013, 191, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Myklebust, J.H.; Irish, J.M.; Brody, J.; Czerwinski, D.K.; Houot, R.; Kohrt, H.E.; Timmerman, J.; Said, J.; Green, M.R.; Delabie, J.; et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood 2013, 121, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.S.; Prod’homme, T.; Youssef, S.; Dunn, S.E.; Steinman, L.; Zamvil, S.S. Neither T-helper type 2 nor Foxp3+ regulatory T cells are necessary for therapeutic benefit of atorvastatin in treatment of central nervous system autoimmunity. J. Neuroinflamm. 2014, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Vilchez, S.; Whitmore, L.; Asmussen, H.; Zareno, J.; Horwitz, R.; Newell-Litwa, K. RhoGTPase regulators orchestrate distinct stages of synaptic development. PLoS ONE 2017, 12, e0170464. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Characterization of human ARHGAP10 gene in silico. Int. J. Oncol. 2004, 25, 1201–1206. [Google Scholar] [PubMed]
- Hellstrom, M.; Ericsson, M.; Johansson, B.; Faraz, M.; Anderson, F.; Henriksson, R.; Nilsson, S.K.; Hedman, H. Cardiac hypertrophy and decreased high-density lipoprotein cholesterol in Lrig3-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1045–R1052. [Google Scholar] [CrossRef] [PubMed]
- Abraira, V.E.; Satoh, T.; Fekete, D.M.; Goodrich, L.V. Vertebrate Lrig3-erbb interactions occur in vitro but are unlikely to play a role in Lrig3-dependent inner ear morphogenesis. PLoS ONE 2010, 5, e8981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraira, V.E.; del Rio, T.; Tucker, A.F.; Slonimsky, J.; Keirnes, H.L.; Goodrich, L.V. Cross-repressive interactions between Lrig3 and netrin 1 shape the architecture of the inner ear. Development 2008, 135, 4091–4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, L.; Larsson, J. Normal hematopoietic stem cell function in mice with enforced expression of the hippo signaling effector YAP1. PLoS ONE 2012, 7, e32013. [Google Scholar] [CrossRef] [PubMed]
- Hoshiba, T.; Otaki, T.; Nemoto, E.; Maruyama, H.; Tanaka, M. Blood-compatible polymer for hepatocyte culture with high hepatocyte-specific functions toward bioartificial liver development. ACS Appl. Mater. Interfaces 2015, 7, 18096–18103. [Google Scholar] [CrossRef] [PubMed]
- Loke, S.Y.; Wong, P.T.; Ong, W.Y. Global gene expression changes in the prefrontal cortex of rabbits with hypercholesterolemia and/or hypertension. Neurochem. Int. 2017, 102, 33–56. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Taniwaki, T.; Kaikoi, Y.; Yamazaki, T. Protective role of the endoplasmic reticulum protein mitsugumin23 against ultraviolet C-induced cell death. FEBS Lett. 2013, 587, 1299–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, J.; Hahnewald, R. Molybdenum cofactor deficiency: Mutations in GPHN, MOCS1, and MOCS2. Hum. Mutat. 2011, 32, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Krizowski, S.; Fischer-Schrader, K.; Niks, D.; Tejero, J.; Sparacino-Watkins, C.; Wang, L.; Ragireddy, V.; Frizzell, S.; Kelley, E.E.; et al. Sulfite oxidase catalyzes single-electron transfer at molybdenum domain to reduce nitrite to nitric oxide. Antioxid. Redox Signal. 2015, 23, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.D.; Bates, W.R.; Reid, S.D. The effects of acute waterborne exposure to sublethal concentrations of molybdenum on the stress response in rainbow trout, oncorhynchus mykiss. PLoS ONE 2015, 10, e0115334. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.; Uetani, N.; Hendriks, W.; Tremblay, M.L.; Bouchard, M. Inactivation of LAR family phosphatase genes Ptprs and Ptprf causes craniofacial malformations resembling pierre-robin sequence. Development 2013, 140, 3413–3422. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Shen, J.C.; Zheng, Z.M.; Harris, C.C. Novel splice variants of ing4 and their possible roles in the regulation of cell growth and motility. J. Biol. Chem. 2006, 281, 34677–34686. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.J.; Siebzehnrubl, F.A.; Schildts, M.J.; Yachnis, A.T.; Smith, G.M.; Smith, A.A.; Scheffler, B.; Reynolds, B.A.; Silver, J.; Steindler, D.A. Chondroitin sulfate proteoglycans potently inhibit invasion and serve as a central organizer of the brain tumor microenvironment. J. Neurosci. 2013, 33, 15603–15617. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, J.; Choi, C. Evaluation of drug-targetable genes by defining modes of abnormality in gene expression. Sci. Rep. 2015, 5, 13576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desh, H.; Gray, S.L.; Horton, M.J.; Raoul, G.; Rowlerson, A.M.; Ferri, J.; Vieira, A.R.; Sciote, J.J. Molecular motor MYO1C, acetyltransferase KAT6B and osteogenetic transcription factor RUNX2 expression in human masseter muscle contributes to development of malocclusion. Arch. Oral Biol. 2014, 59, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, T.; An, D.; Witczak, C.A.; Koh, H.J.; Hirshman, M.F.; Fujii, N.; Goodyear, L.J. Myo1c regulates glucose uptake in mouse skeletal muscle. J. Biol. Chem. 2011, 286, 4133–4140. [Google Scholar] [CrossRef] [PubMed]
- Akahane, K.; Inukai, T.; Zhang, X.; Hirose, K.; Kuroda, I.; Goi, K.; Honna, H.; Kagami, K.; Nakazawa, S.; Endo, K.; et al. Resistance of t-cell acute lymphoblastic leukemia to tumor necrosis factor--related apoptosis-inducing ligand-mediated apoptosis. Exp. Hematol. 2010, 38, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Mao, J.; Yang, Y.; Zhang, Y.; Tian, Y.; Zhu, J. Protective effects of calcitriol on diabetic nephropathy are mediated by down regulation of TGF-β1 and CIP4 in diabetic nephropathy rat. Int. J. Clin. Exp. Pathol. 2015, 8, 3503–3512. [Google Scholar] [PubMed]
- Aulak, K.S.; Davis, A.E., III; Donaldson, V.H.; Harrison, R.A. Chymotrypsin inhibitory activity of normal C1-inhibitor and a P1 arg to his mutant: Evidence for the presence of overlapping reactive centers. Protein Sci. Publ. Protein Soc. 1993, 2, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Imakagura, H.; Futatsumori, M.; Nakayama, K. Recruitment of clathrin onto endosomes by the Tom1-Tollip complex. Biochem. Biophys. Res. Commun. 2006, 341, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Tag | Tissue | Number of Samples | Tag | Tissue | Number of Samples | ||
|---|---|---|---|---|---|---|---|
| Training Dataset | Test Dataset | Training Dataset | Test Dataset | ||||
| T1 | Adipose tissue | 577 | 237 | T2 | Adrenal gland | 145 | 50 |
| T3 | Blood | 511 | 54 | T4 | Blood vessel | 689 | 242 |
| T5 | Brain | 1259 | 455 | T6 | Breast | 214 | 84 |
| T7 | Colon | 345 | 169 | T8 | Esophagus | 686 | 348 |
| T9 | Heart | 412 | 201 | T10 | Liver | 119 | 58 |
| T11 | Lung | 320 | 123 | T12 | Muscle | 430 | 155 |
| T13 | Nerve | 304 | 122 | T14 | Ovary | 97 | 39 |
| T15 | Pancreas | 171 | 82 | T16 | Pituitary | 103 | 82 |
| T17 | Prostate | 106 | 48 | T18 | Skin | 890 | 342 |
| T19 | Small intestine | 88 | 52 | T20 | Spleen | 104 | 60 |
| T21 | Stomach | 192 | 75 | T22 | Testis | 172 | 91 |
| T23 | Thyroid | 323 | 139 | T24 | Uterus | 83 | 32 |
| T25 | Vagina | 96 | 27 | Total | - | 8436 | 3367 |
| Rank | Gene | Description | The Human Protein Atlas [8] | Expression Atlas of EMBL-EBI [37] |
|---|---|---|---|---|
| 1 | ARAF | A-Raf Proto-Oncogene, Serine/Threonine Kinase | Expressed in all | Multiple tissues |
| 2 | ITGA3 | Integrin Subunit Alpha 3 | Mixed | Multiple tissues |
| 3 | SLAIN2 | SLAIN Motif Family Member 2 | Expressed in all | Multiple tissues |
| 4 | ZNF532 | Zinc Finger Protein 532 | Mixed | Multiple tissues |
| 5 | PPIC | Peptidylprolyl Isomerase C | Mixed | Multiple tissues |
| 6 | KDELR1 | KDEL Endoplasmic Reticulum Protein Retention Receptor 1 | Expressed in all | Multiple tissues |
| 7 | NBL1 | Neuroblastoma 1, DAN Family BMP Antagonist | Expressed in all | Multiple tissues |
| 8 | PLP2 | Proteolipid Protein 2 | Expressed in all | Multiple tissues |
| 9 | STAT6 | Signal Transducer and Activator of Transcription 6 | Expressed in all | Multiple tissues |
| 10 | ARHGAP23 | Rho GTPase Activating Protein 23 | Mixed | Multiple tissues |
| 11 | LRIG3 | Leucine Rich Repeats And Immunoglobulin Like Domains 3 | Tissue enhanced (thyroid gland) | Multiple tissues |
| 12 | MANBAL | Mannosidase Beta Like | Expressed in all | Multiple tissues |
| 13 | PTPRA | Protein Tyrosine Phosphatase, Receptor Type A | Expressed in all | Multiple tissues |
| 14 | YAP1 | Yes Associated Protein 1 | Mixed | Multiple tissues |
| 15 | CLIC1 | Chloride Intracellular Channel 1 | Expressed in all | Multiple tissues |
| 16 | TMEM109 | Transmembrane Protein 109 | Expressed in all | Multiple tissues |
| 17 | MOCS2 | Molybdenum Cofactor Synthesis 2 | Expressed in all | Multiple tissues |
| 18 | PTPRF | Protein Tyrosine Phosphatase, Receptor Type F | Mixed | Multiple tissues |
| 19 | MYO1C | Myosin IC | Expressed in all | Multiple tissues |
| 20 | FAM127B | Family with Sequence Similarity 127 Member B | Expressed in all | Multiple tissues |
| 21 | TRIP10 | Thyroid Hormone Receptor Interactor 10 | Expressed in all | Multiple tissues |
| 22 | SERPING1 | Serpin Family G Member 1 | Expressed in all | Multiple tissues |
| 23 | TOM1L2 | Target of Myb1 Like 2 Membrane Trafficking Protein | Expressed in all | Multiple tissues |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chen, L.; Zhang, Y.-H.; Kong, X.; Huang, T.; Cai, Y.-D. A Computational Method for Classifying Different Human Tissues with Quantitatively Tissue-Specific Expressed Genes. Genes 2018, 9, 449. https://doi.org/10.3390/genes9090449
Li J, Chen L, Zhang Y-H, Kong X, Huang T, Cai Y-D. A Computational Method for Classifying Different Human Tissues with Quantitatively Tissue-Specific Expressed Genes. Genes. 2018; 9(9):449. https://doi.org/10.3390/genes9090449
Chicago/Turabian StyleLi, JiaRui, Lei Chen, Yu-Hang Zhang, XiangYin Kong, Tao Huang, and Yu-Dong Cai. 2018. "A Computational Method for Classifying Different Human Tissues with Quantitatively Tissue-Specific Expressed Genes" Genes 9, no. 9: 449. https://doi.org/10.3390/genes9090449
APA StyleLi, J., Chen, L., Zhang, Y.-H., Kong, X., Huang, T., & Cai, Y.-D. (2018). A Computational Method for Classifying Different Human Tissues with Quantitatively Tissue-Specific Expressed Genes. Genes, 9(9), 449. https://doi.org/10.3390/genes9090449