Bioinformatics Tools and Benchmarks for Computational Docking and 3D Structure Prediction of RNA-Protein Complexes


Abstract
1. Introduction
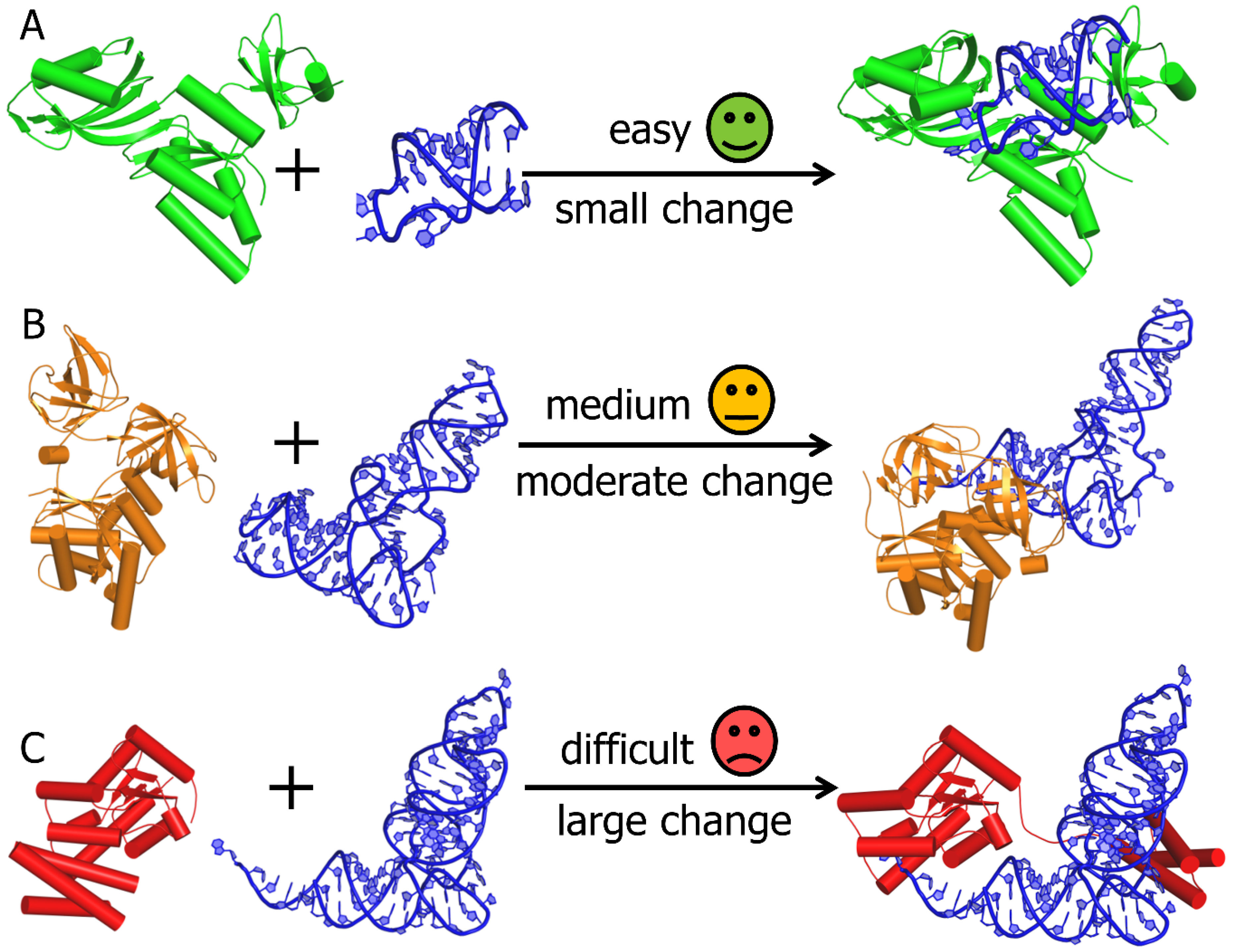
2. Computational Modelling of RNA-Protein Complex Structures
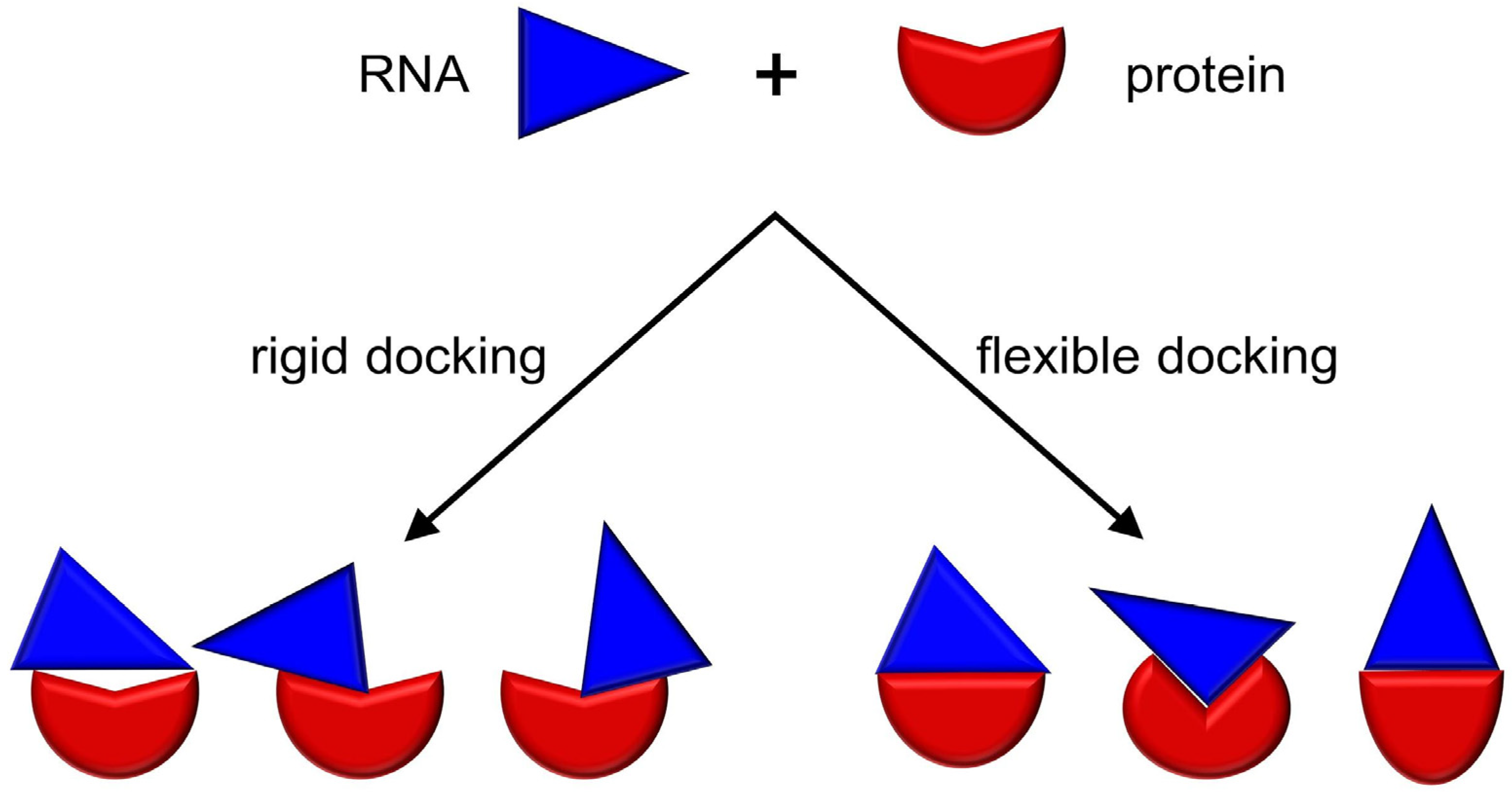
3. RNP Docking Methods (Conformational Sampling with or without Scoring)
4. Other Methods for Three Dimensional Structure Prediction of RNP Complexes
5. Standalone Scoring Methods for RNA-Protein Complexes
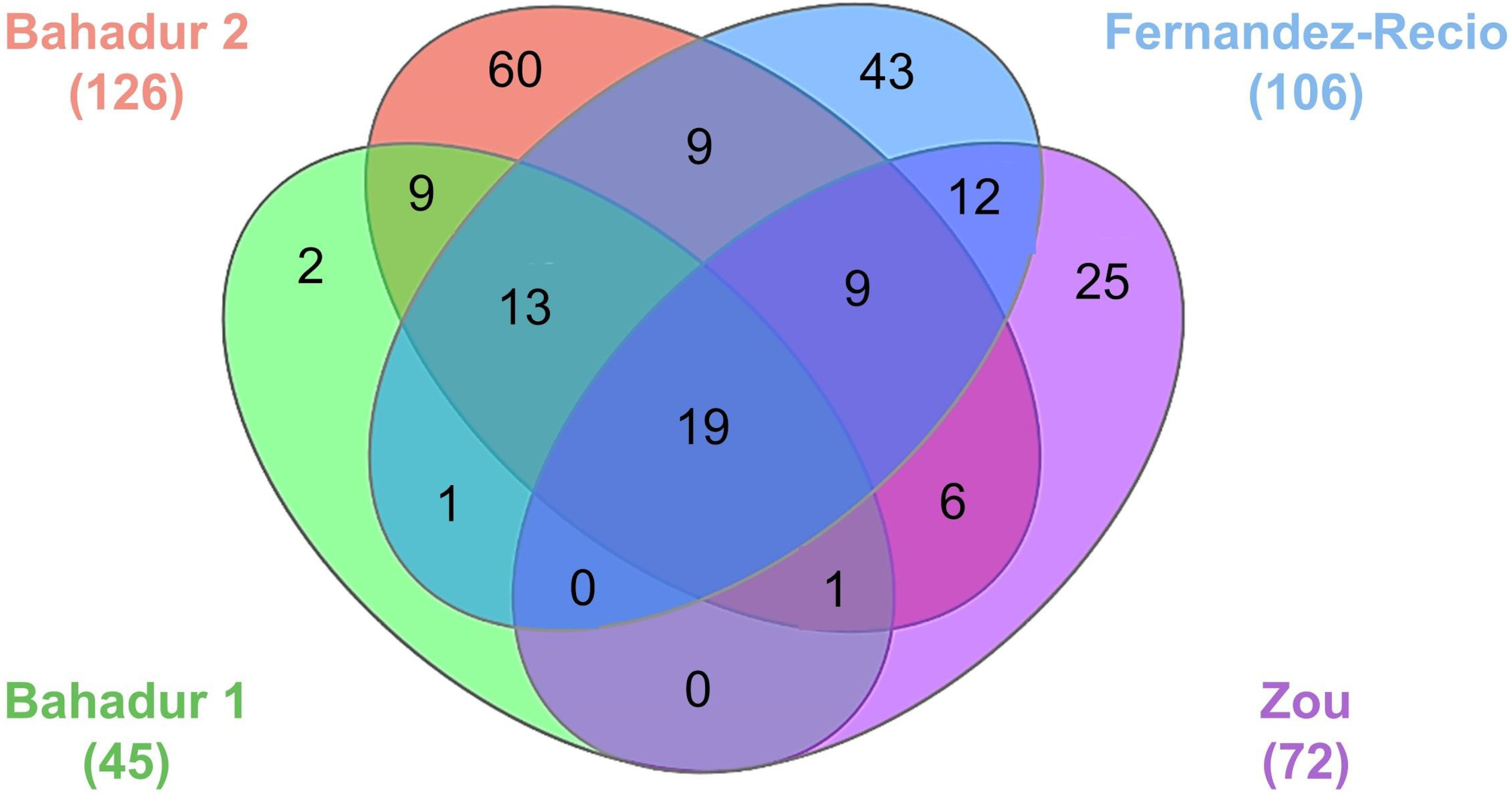
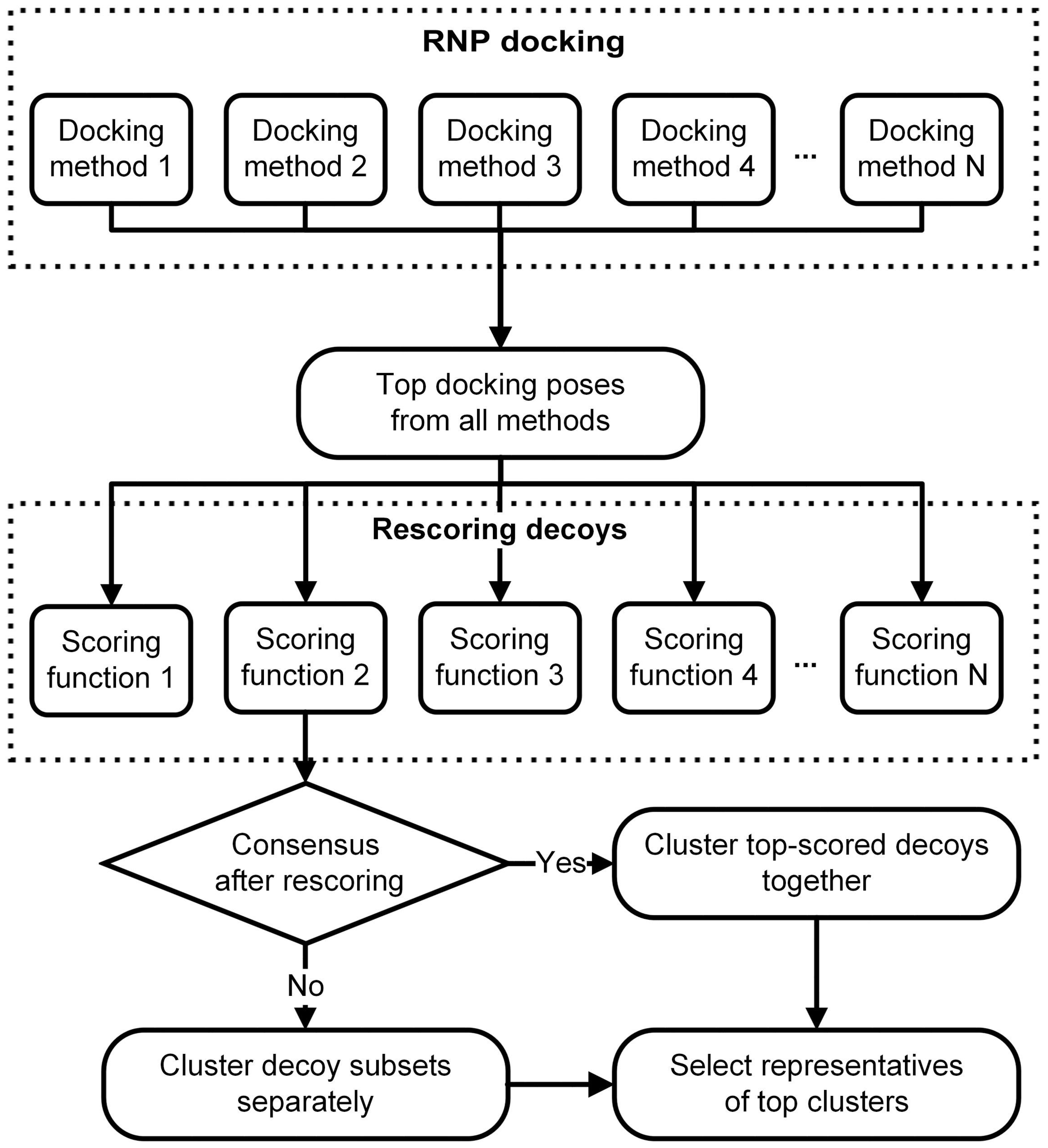
6. RNA-Protein Three-Dimensional Structure Datasets for Benchmarking the Computational Docking Methods and Their Applications
7. Datasets for Ribonucleic Acid-Protein Binding Affinity Prediction and Their Applications
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. RNA Worlds: From Life’s Origins to Diversity in Gene Regulation. Q. Rev. Biol. 2012, 87, 66. [Google Scholar] [CrossRef]
- Chen, Y.; Varani, G. Protein families and RNA recognition. FEBS J. 2005, 272, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F. Ribosomal RNA and translation. Annu. Rev. Biochem. 1991, 60, 191–227. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, C. Messenger RNA splicing in yeast: Clues to why the spliceosome is a ribonucleoprotein. Science 1991, 253, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Reichert, V.L.; Le Hir, H.; Jurica, M.S.; Moore, M.J. 5′ exon interactions within the human spliceosome establish a framework for exon junction complex structure and assembly. Genes Dev. 2002, 16, 2778–2791. [Google Scholar] [CrossRef] [PubMed]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Johnson JM, E. al Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144. [Google Scholar] [CrossRef] [PubMed]
- Minvielle-Sebastia, L.; Keller, W. mRNA polyadenylation and its coupling to other RNA processing reactions and to transcription. Curr. Opin. Cell Biol. 1999, 11, 352–357. [Google Scholar] [CrossRef]
- Valente, L.; Nishikura, K. ADAR Gene Family and A-to-I RNA Editing: Diverse Roles in Posttranscriptional Gene Regulation. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16096031/ (accessed on 21 July 2018).
- Hogg, J.R.; Collins, K. Structured non-coding RNAs and the RNP Renaissance. Curr. Opin. Chem. Biol. 2008, 12, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, B.M.; Brogie, J.E.; Price, D.H. 7SK snRNA: A noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA 2012, 3, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Swain, A.; Misulovin, Z.; Pherson, M.; Gause, M.; Mihindukulasuriya, K.; Rickels, R.A.; Shilatifard, A.; Dorsett, D. Drosophila TDP-43 RNA-Binding Protein Facilitates Association of Sister Chromatid Cohesion Proteins with Genes, Enhancers and Polycomb Response Elements. PLoS Genet. 2016, 12, e1006331. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Chang, K.-W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Cléry, A.-T.; Allain, F.H. From Structure to function of RNA binding domains. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Maris, C.; Dominguez, C.; Allain, F.H.-T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Matunis, M.J.; Matthew Michael, W.; Dreyfuss, G. The pre-mRNA binding K protein contains a novel evolutionary conserved motif. Nucleic Acids Res. 1993, 21, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Ryter, J.M.; Schultz, S.C. Molecular basis of double-stranded RNA-protein interactions: Structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998, 17, 7505–7513. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Searles, M.A.; Klug, A. Crystal structure of a zinc-finger-RNA complex reveals two modes of molecular recognition. Nature 2003, 426, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 2008, 9, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Sperling, J.; Azubel, M.; Sperling, R. Structure and function of the Pre-mRNA splicing machine. Structure 2008, 16, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Matunis, E.L.; Dreyfuss, G. The multiple RNA-binding domains of the mRNA poly(A)-binding protein have different RNA-binding activities. Mol. Cell. Biol. 1991, 11, 3419–3424. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.; Doudna, J.A. Crystallization of RNA and RNA-protein complexes. Methods 2004, 34, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.G.; Hennig, M. RNA Structure Determination by NMR. Methods Mol. Biol. 2008, 452, 29–61. [Google Scholar] [PubMed]
- Garman, E.F. Developments in X-ray crystallographic structure determination of biological macromolecules. Science 2014, 343, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Lapinaite, A.; Simon, B.; Skjaerven, L.; Rakwalska-Bange, M.; Gabel, F.; Carlomagno, T. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature 2013, 502, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Duss, O.; Yulikov, M.; Jeschke, G.; Allain, F.H.-T. EPR-aided approach for solution structure determination of large RNAs or protein-RNA complexes. Nat. Commun. 2014, 5, 3669. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.R.; Chojnowski, G.; Koul, A.; McKenna, S.A.; Bujnicki, J.M. Structural studies of RNA-protein complexes: A hybrid approach involving hydrodynamics, scattering, and computational methods. Methods 2017, 118-119, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.B.; Kazan, H.; Zuberi, K.; Morris, Q.; Hughes, T.R. RBPDB: A database of RNA-binding specificities. Nucleic Acids Res. 2011, 39, D301–D308. [Google Scholar] [CrossRef] [PubMed]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Hentze, M.W.; Preiss, T. Metabolic Enzymes Enjoying New Partnerships as RNA-Binding Proteins. Trends Endocrinol. Metab. 2015, 26, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, Y.; Janga, S.C.; Kao, C.C.; Zhou, Y. Prediction and validation of the unexplored RNA-binding protein atlas of the human proteome. Proteins 2014, 82, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Sowdhamini, R. Genome-wide survey of putative RNA-binding proteins encoded in the human proteome. Mol. Biosyst. 2016, 12, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Murugavel, P.; Sowdhamini, R. hRBPome: A central repository of all known human RNA-binding proteins. BioRxiv 2018. [Google Scholar] [CrossRef]
- Liao, Y.; Castello, A.; Fischer, B.; Leicht, S.; Föehr, S.; Frese, C.K.; Ragan, C.; Kurscheid, S.; Pagler, E.; Yang, H.; et al. The cardiomyocyte RNA-binding proteome: Links to intermediary metabolism and heart disease. Cell Rep. 2016, 16, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Conrad, T.; Albrecht, A.-S.; de Melo Costa, V.R.; Sauer, S.; Meierhofer, D.; Ørom, U.A. Serial interactome capture of the human cell nucleus. Nat. Commun. 2016, 7, 11212. [Google Scholar] [CrossRef] [PubMed]
- Liepelt, A.; Naarmann-de Vries, I.S.; Simons, N.; Eichelbaum, K.; Föhr, S.; Archer, S.K.; Castello, A.; Usadel, B.; Krijgsveld, J.; Preiss, T.; et al. Identification of RNA-binding proteins in macrophages by interactome capture. Mol. Cell. Proteom. 2016, 15, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.S.; Srinivasan, N.; Sowdhamini, R.; Blundell, T.L. Knowledge-based protein modeling. Crit. Rev. Biochem. Mol. Biol. 1994, 29, 1–68. [Google Scholar] [CrossRef] [PubMed]
- Hardin, C.; Pogorelov, T.V.; Luthey-Schulten, Z. Ab initio protein structure prediction. Curr. Opin. Struct. Biol. 2002, 12, 176–181. [Google Scholar] [CrossRef]
- Tozzini, V. Multiscale modeling of proteins. Acc. Chem. Res. 2010, 43, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Protein-protein docking dealing with the unknown. J. Comput. Chem. 2010, 31, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. RNA structure: Advances and assessment of 3D structure prediction. Annu. Rev. Biophys. 2017, 46, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Seetin, M.G.; Mathews, D.H. RNA Structure Prediction: An Overview of Methods. Methods Mol. Biol. 2012, 905, 99–122. [Google Scholar] [CrossRef] [PubMed]
- Dawson, W.K.; Bujnicki, J.M. Computational modeling of RNA 3D structures and interactions. Curr. Opin. Struct. Biol. 2016, 37, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Kasprzak, J.M.; Tuszynska, I.; Magnus, M.; Szczepaniak, K.; Dawson, W.K.; Bujnicki, J.M. Modeling of Protein–RNA Complex Structures Using Computational Docking Methods. Methods Mol. Biol. 2012, 1414, 353–372. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Pedersen, J.T.; Judson, R.; Fidelis, K. A large-scale experiment to assess protein structure prediction methods. Proteins 1995, 23, ii–v. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.A.; Blanchet, M.-F.; Boniecki, M.; Bujnicki, J.M.; Chen, S.-J.; Cao, S.; Das, R.; Ding, F.; Dokholyan, N.V.; Flores, S.C.; et al. RNA-puzzles: A CASP-like evaluation of RNA three-dimensional structure prediction. RNA 2012, 18, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)-Round XII. Proteins 2018, 86 (Suppl. 1), 7–15. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Adamiak, R.W.; Antczak, M.; Batey, R.T.; Becka, A.J.; Biesiada, M.; Boniecki, M.J.; Bujnicki, J.M.; Chen, S.-J.; Cheng, C.Y.; et al. RNA-puzzles round III: 3D RNA structure prediction of five riboswitches and one ribozyme. RNA 2017, 23, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Hall, D.R.; Kozakov, D. Sampling and scoring: A marriage made in heaven. Proteins 2013, 81, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Magnus, M.; Jonak, K.; Dawson, W.; Bujnicki, J.M. NPDock: a web server for protein–nucleic acid docking. Nucleic Acids Res. 2015, 43, W425–W430. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Bujnicki, J.M. DARS-RNP and QUASI-RNP: New statistical potentials for protein-RNA docking. BMC Bioinform. 2011, 12, 348. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Cao, L.B.; Su, J.G.; Yang, Y.X.; Wang, C.X. A new residue-nucleotide propensity potential with structural information considered for discriminating protein-RNA docking decoys. Proteins 2012, 80, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, S.; Guo, D.; Li, L.; Xiao, Y. A novel protocol for three-dimensional structure prediction of RNA-protein complexes. Sci. Rep. 2013, 3, 1887. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014, 42, e55. [Google Scholar] [CrossRef] [PubMed]
- Bastard, K.; Saladin, A.; Prévost, C. Accounting for large amplitude protein deformation during in silico macromolecular docking. Int. J. Mol. Sci. 2011, 12, 1316–1333. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Wodak, S.J. Docking and scoring protein interactions: CAPRI 2009. Proteins 2010, 78, 3073–3084. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, M. Accounting for conformational changes during protein–protein docking. Curr. Opin. Struct. Biol. 2010, 20, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Henrick, K.; Moult, J.; Ten Eyck, L.; Sternberg, M.J.E.; Vajda, S.; Vakser, I.; Wodak, S.J. CAPRI: A Critical Assessment of PRedicted Interactions. Proteins Struct. Funct. Genet. 2003, 52, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Wodak, S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013, 81, 2082–2095. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, H.; Xiao, Y. Using 3dRPC for RNA–protein complex structure prediction. Biophys. Rep. 2016, 2, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.-D.; Ritchie, D.W. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, W445–W449. [Google Scholar] [CrossRef] [PubMed]
- Arnautova, Y.A.; Abagyan, R.; Totrov, M. Protein-RNA docking using ICM. J. Chem. Theory Comput. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Neveu, E.; Ritchie, D.W.; Popov, P.; Grudinin, S. PEPSI-Dock: A detailed data-driven protein–protein interaction potential accelerated by polar Fourier correlation. Bioinformatics 2016, 32, i693–i701. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, B.; Pons, C.; Fernández-Recio, J. pyDockWEB: A web server for rigid-body protein–protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [PubMed]
- Guilhot-Gaudeffroy, A.; Froidevaux, C.; Azé, J.; Bernauer, J. Protein-RNA complexes and efficient automatic docking: expanding RosettaDock possibilities. PLoS ONE 2014, 9, e108928. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Schindler, C.E.M.; Chauvot de Beauchêne, I.; Zacharias, M. A web interface for easy flexible protein-protein docking with ATTRACT. Biophys. J. 2015, 108, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Alexandre, M.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- A Powerful and Innovative Package for Accurate Protein Structure Predictions; Schrödinger, LLC Prime: New York, NY, USA, 2018.
- Bonvin, A.M.J.J.; Karaca, E.; Kastritis, P.L.; Rodrigues, J.P.G.L.M. Defining distance restraints in HADDOCK. Nat. Protoc. 2018, 13, 1503. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; van Dijk, A.D.J.; Hsu, V.; Boelens, R.; Bonvin, A.M.J.J. Information-driven protein-DNA docking using HADDOCK: It is a matter of flexibility. Nucleic Acids Res. 2006, 34, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Kundrotas, P.J.; Zhu, Z.; Janin, J.; Vakser, I.A. Templates are available to model nearly all complexes of structurally characterized proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 9438–9441. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Kundrotas, P.J.; Vakser, I.A.; Liu, S. Template-Based Modeling of Protein-RNA Interactions. PLoS Comput. Biol. 2016, 12, e1005120. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Mathew, O.K.; Sowdhamini, R. RStrucFam: A web server to associate structure and cognate RNA for RNA-binding proteins from sequence information. BMC Bioinform. 2016, 17, 411. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Krepl, M.; Banáš, P.; Kührová, P.; Zgarbová, M.; Jurečka, P.; Havrila, M.; Otyepka, M. How to understand atomistic molecular dynamics simulations of RNA and protein-RNA complexes? Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Bussi, G.; Krepl, M.; Banáš, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, J.; Hamada, M.; Asai, K.; Kameda, T. Improved Accuracy in RNA–Protein Rigid Body Docking by Incorporating Force Field for Molecular Dynamics Simulation into the Scoring Function. J. Chem. Theory Comput. 2016, 12, 4688–4697. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zeng, R.; Tortorella, M.; Wang, J.; Wang, C. Structural Insight into Inhibition of CsrA-RNA Interaction Revealed by Docking, Molecular Dynamics and Free Energy Calculations. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Karaca, E.; Rodrigues, J.P.G.L.M.; Graziadei, A.; Bonvin, A.M.J.J.; Carlomagno, T. M3: An integrative framework for structure determination of molecular machines. Nat. Methods 2017, 14, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Russel, D.; Lasker, K.; Webb, B.; Velázquez-Muriel, J.; Tjioe, E.; Schneidman-Duhovny, D.; Peterson, B.; Sali, A. Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012, 10, e1001244. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.; Patel, T.R.; Chojnowski, G.; Koul, A.; Dzananovic, E.; McEleney, K.; Bujnicki, J.M.; McKenna, S.A. Characterization of the termini of the West Nile virus genome and their interactions with the small isoform of the 2′ 5′-oligoadenylate synthetase family. J. Struct. Biol. 2015, 190, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Friedrich, S.; Kurgan, L. A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues. Brief. Bioinform. 2016, 17, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. A Large-Scale Assessment of Nucleic Acids Binding Site Prediction Programs. PLoS Comput. Biol. 2015, 11, e1004639. [Google Scholar] [CrossRef] [PubMed]
- Gajda, M.J.; Tuszynska, I.; Kaczor, M.; Bakulina, A.Y.; Bujnicki, J.M. FILTREST3D: discrimination of structural models using restraints from experimental data. Bioinformatics 2010, 26, 2986–2987. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, V.; Blundell, T.L.; Fernández-Recio, J. Efficient restraints for protein-protein docking by comparison of observed amino acid substitution patterns with those predicted from local environment. J. Mol. Biol. 2006, 357, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kortemme, T.; Robertson, T.; Baker, D.; Varani, G. A new hydrogen-bonding potential for the design of protein-RNA interactions predicts specific contacts and discriminates decoys. Nucleic Acids Res. 2004, 32, 5147–5162. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Robertson, T.A.; Varani, G. A knowledge-based potential function predicts the specificity and relative binding energy of RNA-binding proteins. FEBS J. 2007, 274, 6378–6391. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cano, L.; Solernou, A.; Pons, C.; Fern?ndez-Recio, J. Structural prediction of protein-RNA interaction by computational docking with propelsity-based statistical potentials. In Biocomputing 2010; World Scientific: Singapore, 2009; pp. 293–301. ISBN 9789814299473. [Google Scholar]
- Zhao, H.; Yang, Y.; Zhou, Y. Structure-based prediction of RNA-binding domains and RNA-binding sites and application to structural genomics targets. Nucleic Acids Res. 2011, 39, 3017–3025. [Google Scholar] [CrossRef] [PubMed]
- Setny, P.; Zacharias, M. A coarse-grained force field for Protein–RNA docking. Nucleic Acids Res. 2011, 39, 9118–9129. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.C.N.; Pilla, S.P.; Bahadur, R.P. Molecular architecture of protein-RNA recognition sites. J. Biomol. Struct. Dyn. 2015, 33, 2738–2751. [Google Scholar] [CrossRef] [PubMed]
- Treger, M.; Westhof, E. Statistical analysis of atomic contacts at RNA-protein interfaces. J. Mol. Recognit. 2001, 14, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11, 2714–2726. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, S.; Zhang, Y. Identification of near-native structures by clustering protein docking conformations. Proteins Struct. Funct. Bioinf. 2007, 68, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; C, N.; P, M.; Bahadur, R.P. A protein-RNA docking benchmark (I): Nonredundant cases. Proteins 2012, 80, 1866–1871. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Mishra, A.; Bahadur, R.P. PRince: a web server for structural and physicochemical analysis of protein-RNA interface. Nucleic Acids Res. 2012, 40, W440–W444. [Google Scholar] [CrossRef] [PubMed]
- Nithin, C.; Mukherjee, S.; Bahadur, R.P. A non-redundant protein--RNA docking benchmark version 2.0. Proteins Struct. Funct. Bioinf. 2017, 85, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cano, L.; Jiménez-García, B.; Fernández-Recio, J. A protein-RNA docking benchmark (II): Extended set from experimental and homology modeling data. Proteins 2012, 80, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. A nonredundant structure dataset for benchmarking protein-RNA computational docking. J. Comput. Chem. 2013, 34, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wang, J. Optimizing scoring function of protein-nucleic acid interactions with both affinity and specificity. PLoS One 2013, 8, e74443. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cano, L.; Romero-Durana, M.; Fernández-Recio, J. Structural and energy determinants in protein-RNA docking. Methods 2017, 118-119, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. Prediction of nucleic acid binding probability in proteins: A neighboring residue network based score. Nucleic Acids Res. 2015, 43, 5340–5351. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jian, Y.; Wang, H.; Zeng, C.; Zhao, Y. RBind: Computational network method to predict RNA binding sites. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Bahadur, R.P. Hydration of protein–RNA recognition sites. Nucleic Acids Res. 2014, 42, 10148–10160. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Nithin, C.; Karampudi, N.B.R.; Mukherjee, S.; Bahadur, R.P. Probing binding hot spots at protein-RNA recognition sites. Nucleic Acids Res. 2016, 44, e9. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bahadur, R.P. An account of solvent accessibility in protein-RNA recognition. Sci. Rep. 2018, 8, 10546. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yan, C. A graph approach to mining biological patterns in the binding interfaces. J. Comput. Biol. 2017, 24, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, J.; Kameda, T.; Asai, K.; Hamada, M. Analysis of base-pairing probabilities of RNA molecules involved in protein–RNA interactions. Bioinformatics 2013, 29, 2524–2528. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Huang, Y.; Liu, S. The dataset for protein-RNA binding affinity. Protein Sci. 2013, 22, 1808–1811. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, Z.; Zhan, W.; Deng, L. Computational identification of binding energy hot spots in protein–RNA complexes using an ensemble approach. Bioinformatics 2017, 34, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xiong, Y.; Gao, H.; Wei, D.-Q.; Mitchell, J.C.; Zhu, X. dbAMEPNI: A database of alanine mutagenic effects for protein–nucleic acid interactions. Database 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B. mCSM–NA: predicting the effects of mutations on protein–nucleic acids interactions. Nucleic Acids Res. 2017, 45, W241–W246. [Google Scholar] [CrossRef] [PubMed]
- Głów, D.; Pianka, D.; Sulej, A.A.; Kozłowski, Ł. P.; Czarnecka, J.; Chojnowski, G.; Skowronek, K.J.; Bujnicki, J.M. Sequence-specific cleavage of dsRNA by Mini-III RNase. Nucleic Acids Res. 2015, 43, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Głów, D.; Kurkowska, M.; Czarnecka, J.; Szczepaniak, K.; Pianka, D.; Kappert, V.; Bujnicki, J.M.; Skowronek, K.J. Identification of protein structural elements responsible for the diversity of sequence preferences among Mini-III RNases. Sci. Rep. 2016, 6, 38612. [Google Scholar] [CrossRef] [PubMed]
- Smietanski, M.; Werner, M.; Purta, E.; Kaminska, K.H.; Stepinski, J.; Darzynkiewicz, E.; Nowotny, M.; Bujnicki, J.M. Structural analysis of human 2′-O-ribose methyltransferases involved in mRNA cap structure formation. Nat. Commun. 2014, 5, 3004. [Google Scholar] [CrossRef] [PubMed]
- Van Laer, B.; Roovers, M.; Wauters, L.; Kasprzak, J.M.; Dyzma, M.; Deyaert, E.; Kumar Singh, R.; Feller, A.; Bujnicki, J.M.; Droogmans, L.; et al. Structural and functional insights into tRNA binding and adenosine N1-methylation by an archaeal Trm10 homologue. Nucleic Acids Res. 2016, 44, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Maravić, G.; Bujnicki, J.M.; Feder, M.; Pongor, S.; Flögel, M. Alanine-scanning mutagenesis of the predicted rRNA-binding domain of ErmC’ redefines the substrate-binding site and suggests a model for protein-RNA interactions. Nucleic Acids Res. 2003, 31, 4941–4949. [Google Scholar] [CrossRef] [PubMed]
- Purta, E.; Kaminska, K.H.; Kasprzak, J.M.; Bujnicki, J.M.; Douthwaite, S. YbeA is the m3Psi methyltransferase RlmH that targets nucleotide 1915 in 23S rRNA. RNA 2008, 14, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Husain, N.; Obranic, S.; Koscinski, L.; Seetharaman, J.; Babic, F.; Bujnicki, J.M.; Maravic-Vlahovicek, G.; Sivaraman, J. Structural basis for the methylation of A1408 in 16S rRNA by a panaminoglycoside resistance methyltransferase NpmA from a clinical isolate and analysis of the NpmA interactions with the 30S ribosomal subunit. Nucleic Acids Res. 2011, 39, 1903–1918. [Google Scholar] [CrossRef] [PubMed]
- Kurowski, M.A.; Bujnicki, J.M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003, 31, 3305–3307. [Google Scholar] [CrossRef] [PubMed]
- Anashkina, A.A.; Kravatsky, Y.; Kuznetsov, E.; Makarov, A.A.; Adzhubei, A.A. Meta-server for automatic analysis, scoring and ranking of docking models. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.; Rother, M.; Boniecki, M.; Puton, T.; Bujnicki, J.M. RNA and protein 3D structure modeling: Similarities and differences. J. Mol. Model. 2011, 17, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S. Detailed close-ups and the big picture of spliceosomes. Curr. Opin. Struct. Biol. 2008, 18, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.C.; Bernauer, J.; Shin, S.; Zhou, R.; Huang, X. Multiscale modeling of macromolecular biosystems. Brief. Bioinform. 2012, 13, 395–405. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Modified from Protein-Protein Docking Method | Docking Method (Rigid/Flexible) | Availability | References | |
|---|---|---|---|---|---|
| Web Server | Standalone | ||||
| 3dRPC | ✗ | Rigid | ✓ | ✓ | [64,71] |
| ClusPro | ✓ | Rigid | ✓ | ✗ | [59] |
| FTDock | ✓ | Rigid | ✗ | ✓ | [72] |
| GRAMM | ✓ | Rigid | ✓ | ✓ | [73] |
| Hex | ✓ | Rigid | ✓ | ✓ | [74] |
| ICM | ✓ | Rigid | ✗ | ✓ | [75] |
| NPDock | ✗ | Rigid | ✓ | ✗ | [60] |
| PatchDock | ✓ | Rigid | ✓ | ✓ | [76] |
| PEPSI-DOCK | ✓ | Rigid | ✗ | ✓ | [77] |
| pyDock | ✓ | Rigid | ✓ | ✓ | [78] |
| RosettaDock | ✓ | Rigid | ✓ | ✓ | [79] |
| ZDOCK | ✓ | Rigid | ✓ | ✓ | [80] |
| ATTRACT | ✓ | Flexible | ✓ | ✓ | [81] |
| HADDOCK | ✓ | Flexible | ✓ | ✓ | [61,82] |
| HDOCK | ✗ | Flexible | ✓ | ✗ | [83] |
| PIPER | ✓ | Flexible | ✗ | ✓ | [84] |
| Prime | ✓ | Flexible | ✗ | ✓ | [85] |
| Name | Structure Representation | Scoring Method | Decoy Discrimination Threshold (RMSD) | Availability as a Standalone Tool | Reference |
|---|---|---|---|---|---|
| Varani’s H-bonding potential | All-atom | H-bonding potential | <3 Å | ✗ | [105] |
| Varani’s all-atom potential | All-atom | All-atom distance-dependent | <5 Å | ✗ | [106] |
| Fernandez’s potential | Coarse-grained | Pairwise residue-ribonucleotide propensity | <10 Å | ✗ | [107] |
| dRNA | All-atom | Volume-fraction corrected DFIRE energy function | NA * | ✗ | [108] |
| DARS-RNP and QUASI-RNP | Coarse-grained | Quasi-chemical potential and decoys as the reference state potentials | <10–15 Å | ✓ | [62] |
| Zacharias’ potential | Coarse-grained | Distance-dependent, coarse-grained force field for protein–RNA interactions. | <8 Å | ✗ | [109] |
| Wang’s potentials | Coarse-grained | Pairwise residue-ribonucleotide propensity with secondary structure information | <10 Å | ✗ | [63] |
| Deck-RP | Coarse-grained | Distance and environment dependent | <15 Å | ✓ | [64] |
| ITScore-PR | All-atom | Pairwise distance dependent atomic interaction potential | <10 Å | ✓ | [65] |
| RPRANK | Coarse-grained | Pairwise residue-nucleotide RMSD | < 10 Å | ✓ | [71] |
| Benchmark | Number of Test Cases | Unbound-Unbound | Unbound-Bound | Bound-Unbound | References |
|---|---|---|---|---|---|
| Bahadur group 1 | 45 | 36 | 9 | 0 | [114] |
| Bahadur group 2 | 126 | 95 | 21 | 10 | [116] |
| Fernandez-Recio group | 106 | 81 | 25 | 0 | [117] |
| Zou group | 72 | 52 | 17 | 3 | [118] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nithin, C.; Ghosh, P.; Bujnicki, J.M. Bioinformatics Tools and Benchmarks for Computational Docking and 3D Structure Prediction of RNA-Protein Complexes. Genes 2018, 9, 432. https://doi.org/10.3390/genes9090432
Nithin C, Ghosh P, Bujnicki JM. Bioinformatics Tools and Benchmarks for Computational Docking and 3D Structure Prediction of RNA-Protein Complexes. Genes. 2018; 9(9):432. https://doi.org/10.3390/genes9090432
Chicago/Turabian StyleNithin, Chandran, Pritha Ghosh, and Janusz M. Bujnicki. 2018. "Bioinformatics Tools and Benchmarks for Computational Docking and 3D Structure Prediction of RNA-Protein Complexes" Genes 9, no. 9: 432. https://doi.org/10.3390/genes9090432
APA StyleNithin, C., Ghosh, P., & Bujnicki, J. M. (2018). Bioinformatics Tools and Benchmarks for Computational Docking and 3D Structure Prediction of RNA-Protein Complexes. Genes, 9(9), 432. https://doi.org/10.3390/genes9090432