Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences


{kind=link}
{kind=link}
Abstract
1. Introduction
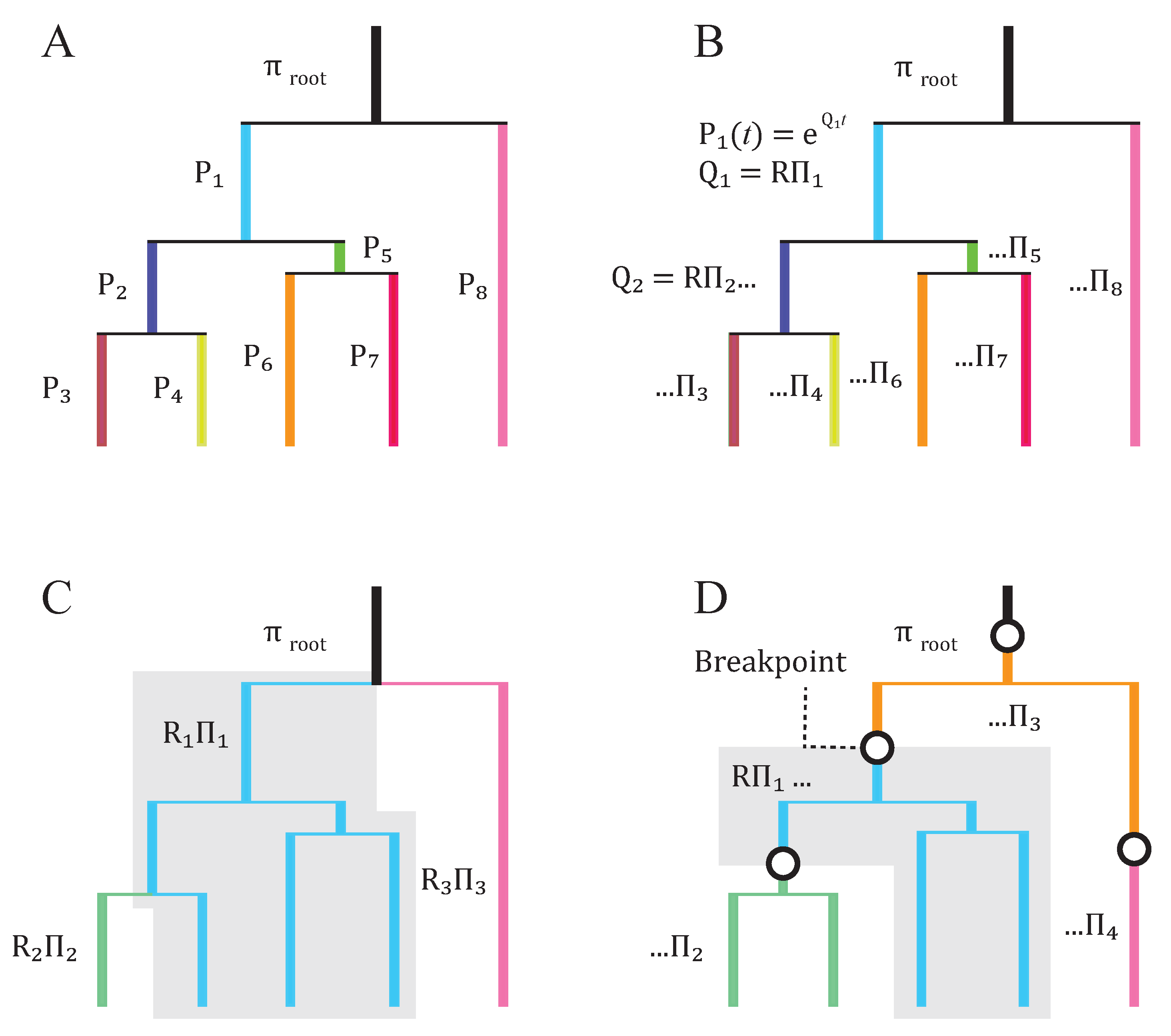
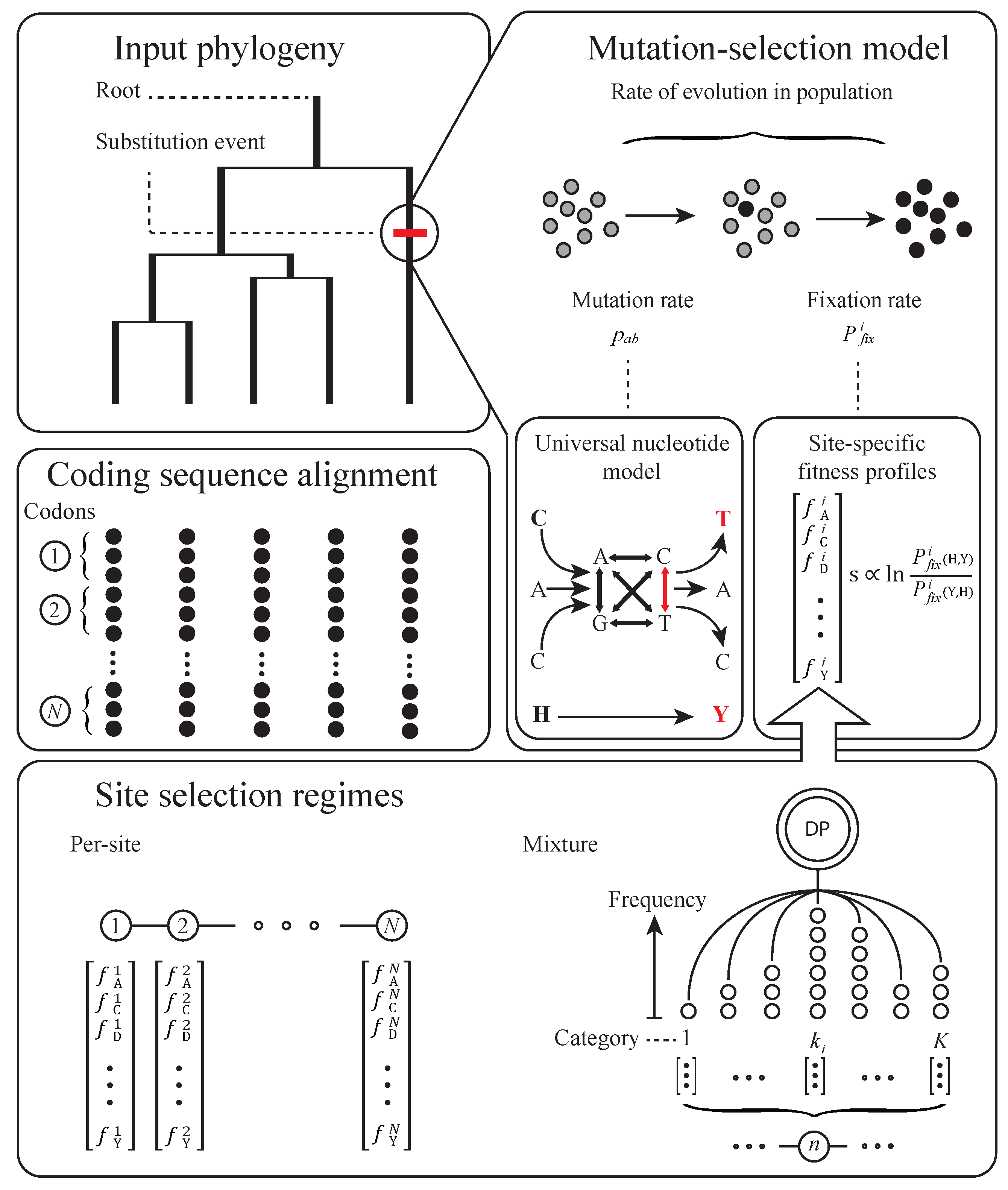
2. Basic Structure of Model
3. Subsequent Implementations and Advances
4. Equilibrium Assumptions and Likelihood
5. Biochemical and Population Genetic Assumptions
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Halpern, A.L.; Bruno, W.J. Evolutionary distances for protein-coding sequences: Modeling site-specific residue frequencies. Mol. Biol. Evol. 1998, 15, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Computational Molecular Evolution; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- O’Brien, J.D.; Minin, V.N.; Suchard, M.A. Learning to count: Robust estimates for labeled distances between molecular sequences. Mol. Biol. Evol. 2009, 26, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.B.; Liberles, D.A. Selection on protein structure, interaction, and sequence. Protein Sci. 2016, 25, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Alberch, P. From genes to phenotype: dynamical systems and evolvability. Genetica 1991, 84, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [PubMed]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [PubMed]
- Thorne, J.L.; Lartillot, N.; Rodrigue, N.; Choi, S.C. Codon models as a vehicle for reconciling population genetics with inter-specific sequence data. In Codon Evolution: Mechanisms and Models; Oxford University Press: Oxford, UK, 2012; pp. 97–110. [Google Scholar]
- Golding, B.; Felsenstein, J. A maximum likelihood approach to the detection of selection from a phylogeny. J. Mol. Evol. 1990, 31, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evol. 2008, 25, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. On the probability of fixation of mutant genes in a population. Genetics 1962, 47, 713–719. [Google Scholar] [PubMed]
- Sella, G.; Hirsh, A. The application of statistical physics to evolutionary biology. Proc. Natl. Acad. Sci. USA 2005, 102, 9541–9546. [Google Scholar] [CrossRef] [PubMed]
- Krukov, I.; de Sanctis, B.; de Koning, A.P.J. Wright–Fisher exact solver (WFES): Scalable analysis of population genetic models without simulation or diffusion theory. Bioinformatics 2017, 33, 1416–1417. [Google Scholar] [CrossRef] [PubMed]
- De Koning, A.J.; De Sanctis, B.D. The rate of observable molecular evolution when mutation may not be weak. bioRxiv 2018, 259507. [Google Scholar] [CrossRef]
- Jones, D.T. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences1. J. Mol. Biol. 1999, 287, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.M.; Jones, D.T.; Kishino, H.; Goldman, N.; Thorne, J.L. Protein evolution with dependence among codons due to tertiary structure. Mol. Biol. Evol. 2003, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N.; Bryant, D.; Philippe, H. Site interdependence attributed to tertiary structure in amino acid sequence evolution. Gene 2005, 347, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Kleinman, C.L.; Philippe, H.; Lartillot, N. Computational methods for evaluating phylogenetic models of coding sequence evolution with dependence between codons. Mol. Biol. Evol. 2009, 26, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Dos Santos, H.G.; Posada, D.; Bastolla, U. Protein evolution along phylogenetic histories under structurally constrained substitution models. Bioinformatics 2013, 29, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Weber, C.C.; Liberles, D.A.; Bastolla, U. ProtASR: An evolutionary framework for ancestral protein reconstruction with selection on folding stability. Syst. Biol. 2017, 66, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Sánchez-Cobos, A.; Bastolla, U. Maximum-likelihood phylogenetic inference with selection on protein folding stability. Mol. Biol. Evol. 2015, 32, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- De Koning, A.J.; Gu, W.; Pollock, D.D. Rapid likelihood analysis on large phylogenies using partial sampling of substitution histories. Mol. Biol. Evol. 2009, 27, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Spielman, S.J.; Wilke, C.O. The relationship between dN/dS and scaled selection coefficients. Mol. Biol. Evol. 2015, 32, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Philippe, H.; Lartillot, N. Mutation-selection models of coding sequence evolution with site-heterogeneous amino acid fitness profiles. Proc. Natl. Acad. Sci. USA 2010, 107, 4629–4634. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N. Site-heterogeneous mutation-selection models within the PhyloBayes-MPI package. Bioinformatics 2013, 30, 1020–1021. [Google Scholar] [CrossRef] [PubMed]
- Tamuri, A.U.; dos Reis, M.; Goldstein, R.A. Using site-wise mutation-selection models to estimate the distribution of selection coefficients from phylogenetic data. Genetics 2011, 111. [Google Scholar] [CrossRef]
- Grahnen, J.A.; Nandakumar, P.; Kubelka, J.; Liberles, D.A. Biophysical and structural considerations for protein sequence evolution. BMC Evol. Biol. 2011, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N. On the statistical interpretation of site-specific variables in phylogeny-based substitution models. Genetics 2012. [Google Scholar] [CrossRef] [PubMed]
- Tamuri, A.U.; Goldman, N.; dos Reis, M. A penalized-likelihood method to estimate the distribution of selection coefficients from phylogenetic data. Genetics 2014, 197, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Spielman, S.J.; Wilke, C.O. Extensively parameterized mutation–selection models reliably capture site-specific selective constraint. Mol. Biol. Evol. 2016, 33, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol. Biol. Evol. 2014, 31, 1956–1978. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. An experimentally informed evolutionary model improves phylogenetic fit to divergent lactamase homologs. Mol. Biol. Evol. 2014, 31, 2753–2769. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. Identification of positive selection in genes is greatly improved by using experimentally informed site-specific models. Biol. Direct 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N. Detecting adaptation in protein-coding genes using a Bayesian site-heterogeneous mutation-selection codon substitution model. Mol. Biol. Evol. 2017, 34, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Gouy, M. Inferring pattern and process: Maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol. Biol. Evol. 1998, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.; Hartigan, J.A. Statistical analysis of hominoid molecular evolution. Stat. Sci. 1987, 2, 191–207. [Google Scholar] [CrossRef]
- Chang, J.T. Full reconstruction of Markov models on evolutionary trees: Identifiability and consistency. Math. Biosci. 1996, 137, 51–73. [Google Scholar] [CrossRef]
- Zou, L.; Susko, E.; Field, C.; Roger, A.J. The parameters of the Barry and Hartigan general Markov model are statistically nonIdentifiable. Syst. Biol. 2011, 60, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, B.D.; Yap, V.B.; Zhang, R.L.; Huttley, G.A. Genetic distance for a general non-stationary Markov substitution process. Syst. Biol. 2015, 64, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Roberts, D. On the use of nucleic acid sequences to infer early branchings in the tree of life. Mol. Biol. Evol. 1995, 12, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, S.; Lartillot, N. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol. Biol. Evol. 2006, 23, 2058–2071. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Boussau, B.; Gouy, M. A branch-heterogeneous model of protein evolution for efficient inference of ancestral sequences. Syst. Biol. 2013, 62, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Foster, P. Modeling compositional heterogeneity. Syst. Biol. 2004, 53, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Gowri-Shankar, V.; Rattray, M. A reversible jump method for Bayesian phylogenetic inference with a nonhomogeneous substitution model. Mol. Biol. Evol. 2007, 24, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, S.; Lartillot, N. A site- and time-heterogeneous model of amino acid replacement. Mol. Biol. Evol. 2008, 25, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Shore, J.A.; Sumner, J.G.; Holland, B.R. Closed codon models: Just a hopeless dream? arXiv, 2018; arXiv:1804.11249. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA-sequences—A maximum-likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Boussau, B.; Gouy, M. Efficient likelihood computations with nonreversible models of evolution. Syst. Biol. 2006, 55, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.W.; Susko, E.; Field, C.; Roger, A.J. Fitting nonstationary general-time-reversible models to obtain edge-lengths and frequencies for the Barry-Hartigan model. Syst. Biol. 2012, 61, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and effects of N-terminal codon bias in bacterial genes. Science 2013, 1241934. [Google Scholar] [CrossRef] [PubMed]
- Bentele, K.; Saffert, P.; Rauscher, R.; Ignatova, Z.; Blüthgen, N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wu, W.B.; Comeron, J.M.; Kreitman, M.; Li, W.H. Intragenic spatial patterns of codon usage bias in prokaryotic and eukaryotic genomes. Genetics 2004, 168, 2245–2260. [Google Scholar] [CrossRef] [PubMed]
- Hockenberry, A.J.; Sirer, M.I.; Amaral, L.A.N.; Jewett, M.C. Quantifying position-dependent codon usage bias. Mol. Biol. Evol. 2014, 31, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.S.; Barral, J.M. Genetic code redundancy and its influence on the encoded polypeptides. Comput. Struct. Biotechnol. J. 2012, 1, e201204006. [Google Scholar] [CrossRef] [PubMed]
- Pouyet, F.; Bailly-Bechet, M.; Mouchiroud, D.; Guéguen, L. SENCA: A multilayered codon model to study the origins and dynamics of codon usage. Gen. Biol. Evol. 2016, 8, 2427–2441. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Lartillot, N.; Philippe, H. Bayesian comparisons of codon substitution models. Genetics 2008, 180, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue, N.; Philippe, H. Mechanistic revisions of phenomenological modeling strategies in molecular evolution. Trend. Genet. 2010, 26, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Liberles, D.A.; Tisdell, M.D.; Grahnen, J.A. Binding constraints on the evolution of enzymes and signalling proteins: The important role of negative pleiotropy. Proc. R. Soc. Lond. B Biol. Sci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Echave, J.; Wilke, C.O. Biophysical models of protein evolution: understanding the patterns of evolutionary sequence divergence. Ann. Rev. Biophys. 2017, 46, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Pollock, D.D.; Thiltgen, G.; Goldstein, R.A. Amino acid coevolution induces an evolutionary Stokes shift. Proc. Natl. Acad. Sci. USA 2012, 109, E1352–E1359. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; McCandlish, D.M.; Plotkin, J.B. Contingency and entrenchment in protein evolution under purifying selection. Proc. Natl. Acad. Sci. USA 2015, 112, E3226–E3235. [Google Scholar] [CrossRef] [PubMed]
- Platt, A.; Weber, C.C.; Liberles, D.A. Protein evolution depends on multiple distinct population size parameters. BMC Evol. Biol. 2018, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Liberles, D.A.; Teufel, A.I.; Liu, L.; Stadler, T. On the need for mechanistic models in computational genomics and metagenomics. Gen. Biol. Evol. 2013, 5, 2008–2018. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teufel, A.I.; Ritchie, A.M.; Wilke, C.O.; Liberles, D.A. Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences. Genes 2018, 9, 409. https://doi.org/10.3390/genes9080409
Teufel AI, Ritchie AM, Wilke CO, Liberles DA. Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences. Genes. 2018; 9(8):409. https://doi.org/10.3390/genes9080409
Chicago/Turabian StyleTeufel, Ashley I., Andrew M. Ritchie, Claus O. Wilke, and David A. Liberles. 2018. "Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences" Genes 9, no. 8: 409. https://doi.org/10.3390/genes9080409
APA StyleTeufel, A. I., Ritchie, A. M., Wilke, C. O., & Liberles, D. A. (2018). Using the Mutation-Selection Framework to Characterize Selection on Protein Sequences. Genes, 9(8), 409. https://doi.org/10.3390/genes9080409