Roles of Mitochondrial DNA Mutations in Stem Cell Ageing


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Somatic mtDNA Mutations in Normal Ageing Humans
3. Ageing of Somatic Stem Cell Populations in mtDNA-Mutator Mice
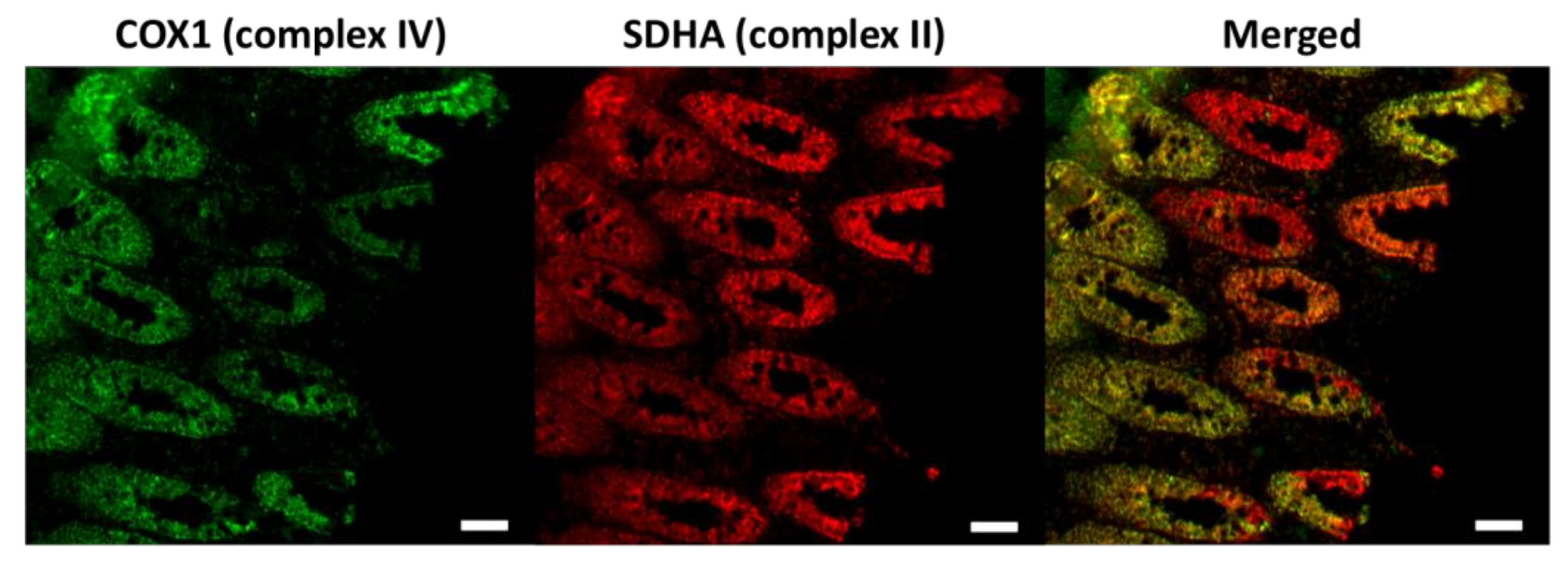
3.1. Age-Dependent Dysfunction of Stem Cells and Their Progenitors in Mitotic Tissue
3.2. Ageing of Progenitor Cells in Post-Mitotic Tissue
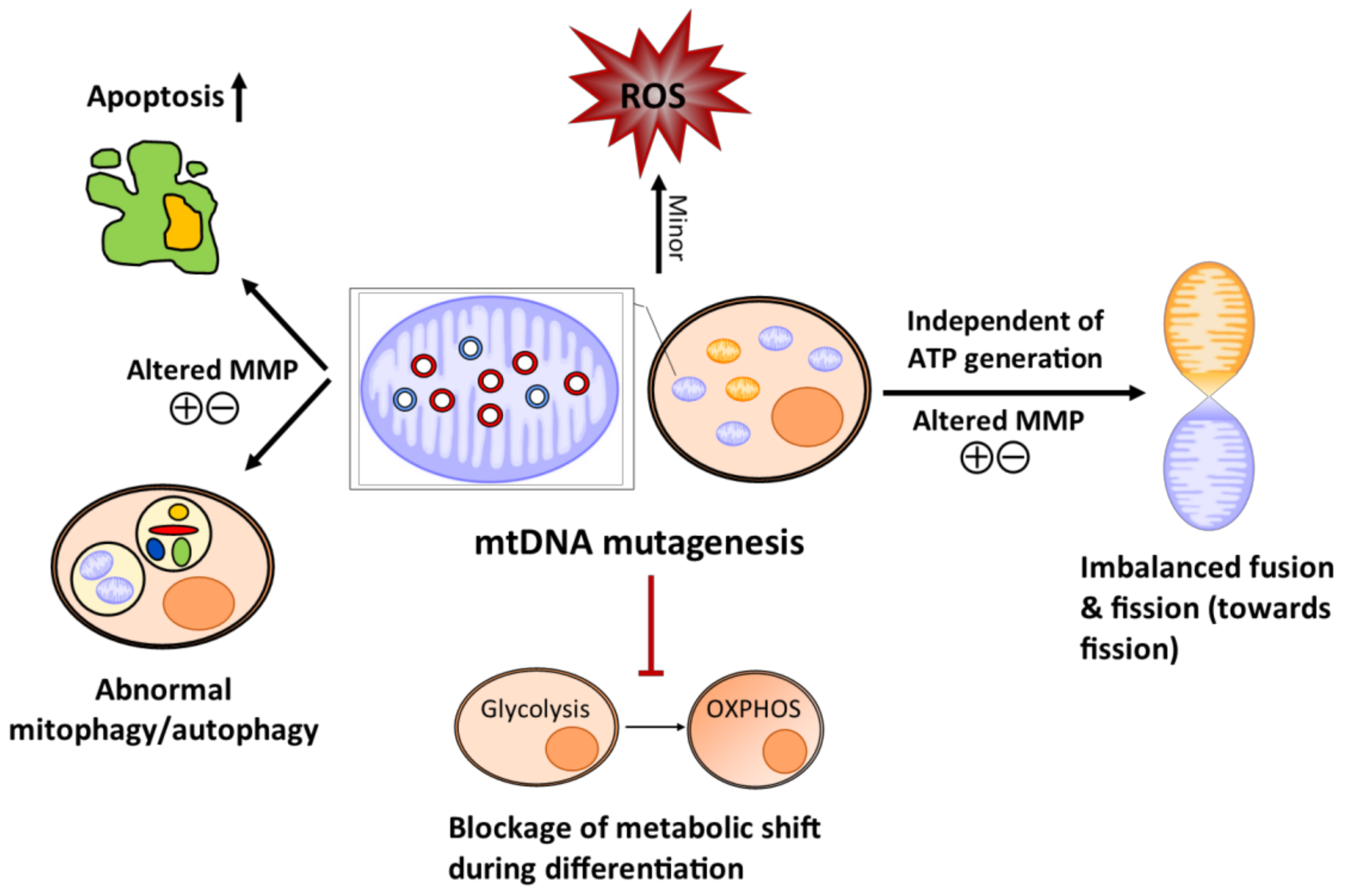
4. Potential Mechanism Whereby Age-Related mtDNA Mutagenesis Affects Somatic Stem Cells
5. Behaviour of mtDNA Mutations Inherited through Germline with Age
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rando, T.A. Stem cells, ageing and the quest for immortality. Nature 2006, 441, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Rando, T.A. Emerging models and paradigms for stem cell ageing. Nat. Cell Biol. 2011, 13, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rando, T.A. Manifestations and mechanisms of stem cell aging. J. Cell Biol. 2011, 193, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Rudnicki, M.A. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 2000, 218, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Ming, G.-L.; Song, H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.; Clayton, D.A. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 1977, 11, 719–727. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.-G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Linnane, A.; Ozawa, T.; Marzuki, S.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 333, 642–645. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Müller-Höcker, J. Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: An age-related alteration. J. Neurol. Sci. 1990, 100, 14–21. [Google Scholar] [CrossRef]
- Müller-Höcker, J. Cytochrome c oxidase deficient cardiomyocytes in the human heart—an age-related phenomenon. A histochemical ultracytochemical study. Am. J. Pathol. 1989, 134, 1167–1173. [Google Scholar] [PubMed]
- Cottrell, D.A.; Blakely, E.L.; Johnson, M.A.; Ince, P.G.; Borthwick, G.M.; Turnbull, D.M. Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol. Aging 2001, 22, 265–272. [Google Scholar] [CrossRef]
- Fellous, T.G.; Islam, S.; Tadrous, P.J.; Elia, G.; Kocher, H.M.; Bhattacharya, S.; Mears, L.; Turnbull, D.M.; Taylor, R.W.; Greaves, L.C.; et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 2009, 49, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.A.C.; Greaves, L.C.; Gutierrez-Gonzalez, L.; Rodriguez-Justo, M.; Deheragoda, M.; Leedham, S.J.; Taylor, R.W.; Lee, C.Y.; Preston, S.L.; Lovell, M.; et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology 2008, 134, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Gonzalez, L.; Deheragoda, M.; Elia, G.; Leedham, S.J.; Shankar, A.; Imber, C.; Jankowski, J.A.; Turnbull, D.M.; Novelli, M.; Wright, N.A.; et al. Analysis of the clonal architecture of the human small intestinal epithelium establishes a common stem cell for all lineages and reveals a mechanism for the fixation and spread of mutations. J. Pathol. 2009, 217, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Investig. 2003, 112, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Barron, M.J.; Plusa, S.; Kirkwood, T.B.; Mathers, J.C.; Taylor, R.W.; Turnbull, D.M. Defects in multiple complexes of the respiratory chain are present in ageing human colonic crypts. Exp. Gerontol. 2010, 45, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.G.; Kajigaya, S.; Tarnowka, M.; McCoy, J.P.; Levin, B.C.; Young, N.S. Mitochondrial DNA sequence heterogeneity in circulating normal human CD34 cells and granulocytes. Blood 2004, 103, 4466–4477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, Y.; Luo, X.; Zhu, J.; Zhang, X.; Zhu, Y.; Cheng, H.; Xia, Z.; Su, N.; Zhang, N.; Zhou, J. Mitochondrial DNA 4977 bp deletion is a common phenomenon in hair and increases with age. Bosn. J. Basic Med. Sci. 2012, 12, 187–192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shin, M.G.; Kajigaya, S.; McCoy, J.P.; Levin, B.C.; Young, N.S. Marked mitochondrial DNA sequence heterogeneity in single CD34+ cell clones from normal adult bone marrow. Blood 2004, 103, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Elson, J.L.; Samuels, D.C.; Turnbull, D.M.; Chinnery, P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001, 68, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Elson, J.L.; Nooteboom, M.; Grady, J.P.; Taylor, G.A.; Taylor, R.W.; Mathers, J.C.; Kirkwood, T.B.L.; Turnbull, D.M. Comparison of mitochondrial mutation spectra in ageing human colonic epithelium and disease: Absence of evidence for purifying selection in somatic mitochondrial DNA point mutations. PLoS Genet. 2012, 8, e1003082. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Taylor, G.; Elson, J.L.; Bender, A.; Taylor, R.W.; Morris, C.M.; Turnbull, D.M. The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell 2009, 8, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Pallotti, F.; Chen, X.; Bonilla, E.; Schon, E.A. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am. J. Hum. Genet. 1996, 59, 591–602. [Google Scholar] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Preston, S.L.; Tadrous, P.J.; Taylor, R.W.; Barron, M.J.; Oukrif, D.; Leedham, S.J.; Deheragoda, M.; Sasieni, P.; Novelli, M.R.; et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc. Natl. Acad. Sci. USA 2006, 103, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Nooteboom, M.; Johnson, R.; Taylor, R.W.; Wright, N.A.; Lightowlers, R.N.; Kirkwood, T.B.L.; Mathers, J.C.; Turnbull, D.M.; Greaves, L.C. Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts. Aging Cell 2010, 9, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Logan, T.D.; Hochberg, M.L.; Shelat, S.G.; Yu, X.; Wilding, G.E.; Tan, W.; Kujoth, G.C.; Prolla, T.A.; Selak, M.A.; et al. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood 2009, 114, 4045–4053. [Google Scholar] [CrossRef] [PubMed]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hämäläinen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Götz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in POLG mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.G.; Magness, S.; Kujoth, G.C.; Prolla, T.A.; Maeda, N. Mitochondrial DNA polymerase editing mutation, POLGD257A, disturbs stem-progenitor cell cycling in the small intestine and restricts excess fat absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G914–G924. [Google Scholar] [CrossRef] [PubMed]
- Baines, H.L.; Stewart, J.B.; Stamp, C.; Zupanic, A.; Kirkwood, T.B.L.; Larsson, N.-G.; Turnbull, D.M.; Greaves, L.C. Similar patterns of clonally expanded somatic mtDNA mutations in the colon of heterozygous mtDNA mutator mice and ageing humans. Mech. Ageing Dev. 2014, 139, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Wredenberg, A.; Cansu, Z.; Trifunovic, A.; Larsson, N.-G. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Orogo, A.M.; Gonzalez, E.R.; Kubli, D.A.; Baptista, I.L.; Ong, S.-B.; Prolla, T.A.; Sussman, M.A.; Murphy, A.N.; Gustafsson, Å.B. Accumulation of mitochondrial DNA mutations disrupts cardiac progenitor cell function and reduces survival. J. Biol. Chem. 2015, 290, 22061–22075. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.Q.; Suda, T. Reactive oxygen species and mitochondrial homeostasis as regulators of stem cell fate and function. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, R.H. Mitochondria and mtDNA integrity in stem cell function and differentiation. Curr. Opin. Gene. Dev. 2016, 38, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [PubMed]
- Baines, H.L.; Turnbull, D.M.; Greaves, L.C. Human stem cell aging: Do mitochondrial DNA mutations have a causal role? Aging Cell 2014, 13, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Leoncini, S.; Pecorelli, A.; Wortmann, S.B.; Ahola, S.; Forsström, S.; Guerranti, R.; De Felice, C.; Smeitink, J.; Ciccoli, L.; et al. mtDNA mutagenesis impairs elimination of mitochondria during erythroid maturation leading to enhanced erythrocyte destruction. Nat. Commun. 2015, 6, 6494. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Kujoth, G.C.; Kim, M.J.; Hacker, T.A.; Vermulst, M.; Weindruch, R.; Prolla, T.A. Effects of calorie restriction on the lifespan and healthspan of POLG mitochondrial mutator mice. PLoS ONE 2017, 12, e0171159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pickrell, A.M.; Rossi, S.G.; Pinto, M.; Dillon, L.M.; Hida, A.; Rotundo, R.L.; Moraes, C.T. Transient systemic mtDNA damage leads to muscle wasting by reducing the satellite cell pool. Hum. Mol. Genet. 2013, 22, 3976–3986. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Pickrell, A.M.; Wang, X.; Bacman, S.R.; Yu, A.; Hida, A.; Dillon, L.M.; Morton, P.D.; Malek, T.R.; Williams, S.L.; et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 2016, 24, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Li-Harms, X.; Milasta, S.; Lynch, J.; Wright, C.; Joshi, A.; Iyengar, R.; Neale, G.; Wang, X.; Wang, Y.-D.; Prolla, T.A.; et al. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood 2015, 125, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Larsson, N.-G.; Holme, E.; Kristiansson, B.; Oldfors, A.; Tulinius, M. Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr. Res. 1990, 28, 131–136. [Google Scholar] [CrossRef] [PubMed]
- McShane, M.A.; Hammans, S.R.; Sweeney, M.; Holt, I.J.; Beattie, T.J.; Brett, E.M.; Harding, A.E. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am. J. Hum. Genet. 1991, 48, 39–42. [Google Scholar] [PubMed]
- Nørby, S.; Lestienne, P.; Nelson, I.; Nielsen, I.M.; Schmalbruch, H.; Sjö, O.; Warburg, M. Juvenile Kearns-Sayre syndrome initially misdiagnosed as a psychosomatic disorder. J. Med. Genet. 1994, 31, 45–50. [Google Scholar] [CrossRef] [PubMed]
- ’t Hart, L.M.; Jansen, J.J.; Lemkes, H.H.; de Knijff, P.; Maassen, J.A. Heteroplasmy levels of a mitochondrial gene mutation associated with diabetes mellitus decrease in leucocyte DNA upon aging. Hum. Mutat. 1996, 7, 193–197. [Google Scholar] [CrossRef]
- Olsson, C.; Johnsen, E.; Nilsson, M.; Wilander, E.; Syvanen, A.C.; Lagerstrom-Fermer, M. The level of the mitochondrial mutation A3243G decreases upon ageing in epithelial cells from individuals with diabetes and deafness. Eur. J. Hum. Genet. 2001, 9, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Poulton, J.; Marchington, D.; Suomalainen, A. Decrease of 3243 A→G mtDNA mutation from blood in MELAS syndrome: A longitudinal study. Am. J. Hum. Genet. 2001, 68, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, A.L.; Andersen, P.H.; Kyvik, K.O.; Jeppesen, T.D.; Vissing, J.; Schwartz, M. Tissue specific distribution of the 3243A→G mtDNA mutation. J. Med. Genet. 2006, 43, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Rajasimha, H.K.; Chinnery, P.F.; Samuels, D.C. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A→G mutation in blood. Am. J. Hum. Genet. 2008, 82, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, J.H.; Baines, H.L.; Bratic, A.; Simard, M.-L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.-G.; Greaves, L.C.; et al. A phenotype-driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Vijg, J. Mitochondrial DNA mutations and aging: Devils in the details? Trends Genet. 2009, 25, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Popadin, K.; Gunbin, K.V.; Khrapko, K. Mitochondrial DNA mutations and cancer: Lessons from the parathyroid. Am. J. Pathol. 2014, 184, 2852–2854. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.-S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2012, 493, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sha, H.; Ji, D.; Zhang, H.L.; Chen, D.; Cao, Y.; Zhu, J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 2014, 157, 1591–1604. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, T.; Turnbull, D.M.; Greaves, L.C. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes 2018, 9, 182. https://doi.org/10.3390/genes9040182
Su T, Turnbull DM, Greaves LC. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes. 2018; 9(4):182. https://doi.org/10.3390/genes9040182
Chicago/Turabian StyleSu, Tianhong, Doug M. Turnbull, and Laura C. Greaves. 2018. "Roles of Mitochondrial DNA Mutations in Stem Cell Ageing" Genes 9, no. 4: 182. https://doi.org/10.3390/genes9040182
APA StyleSu, T., Turnbull, D. M., & Greaves, L. C. (2018). Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes, 9(4), 182. https://doi.org/10.3390/genes9040182