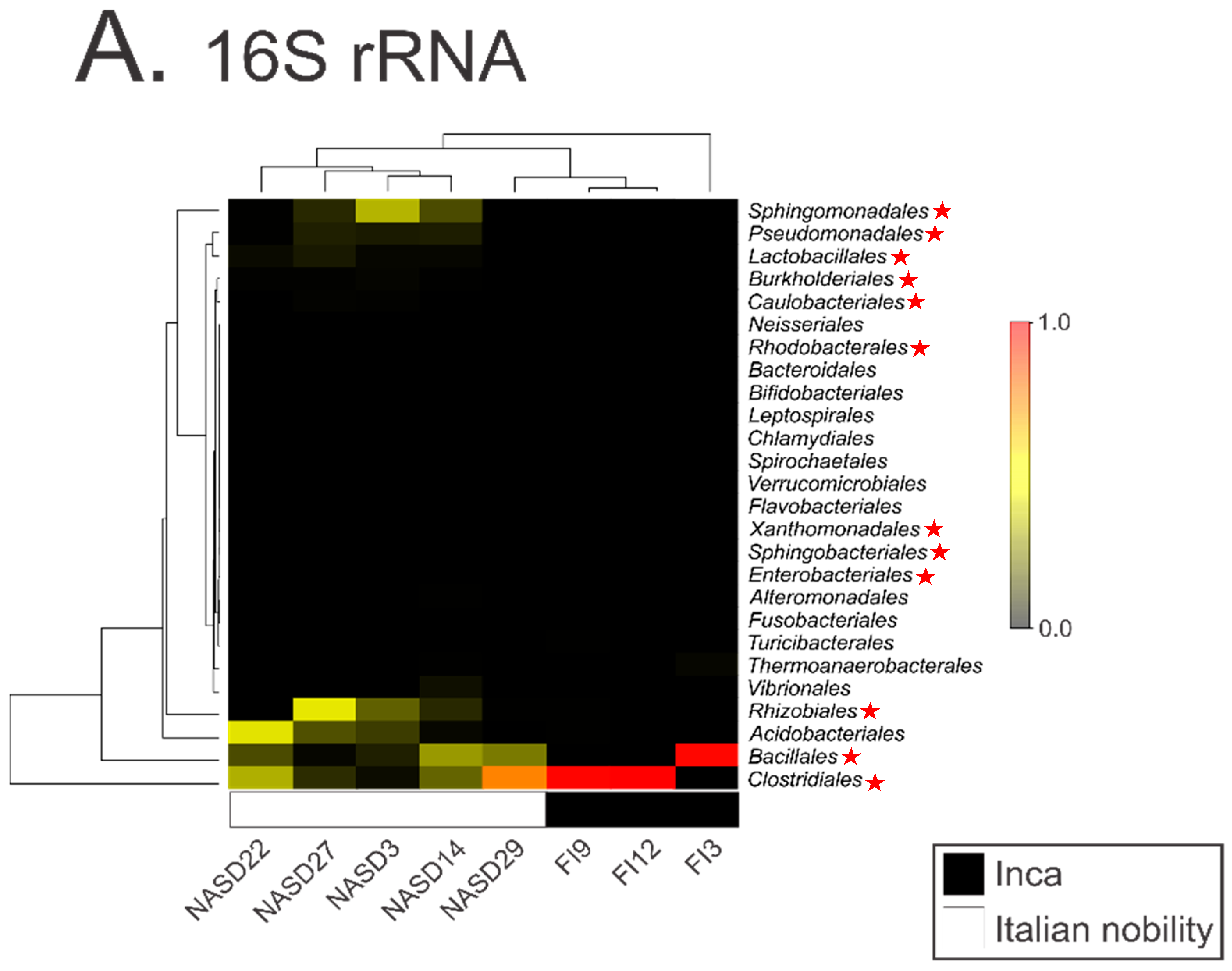
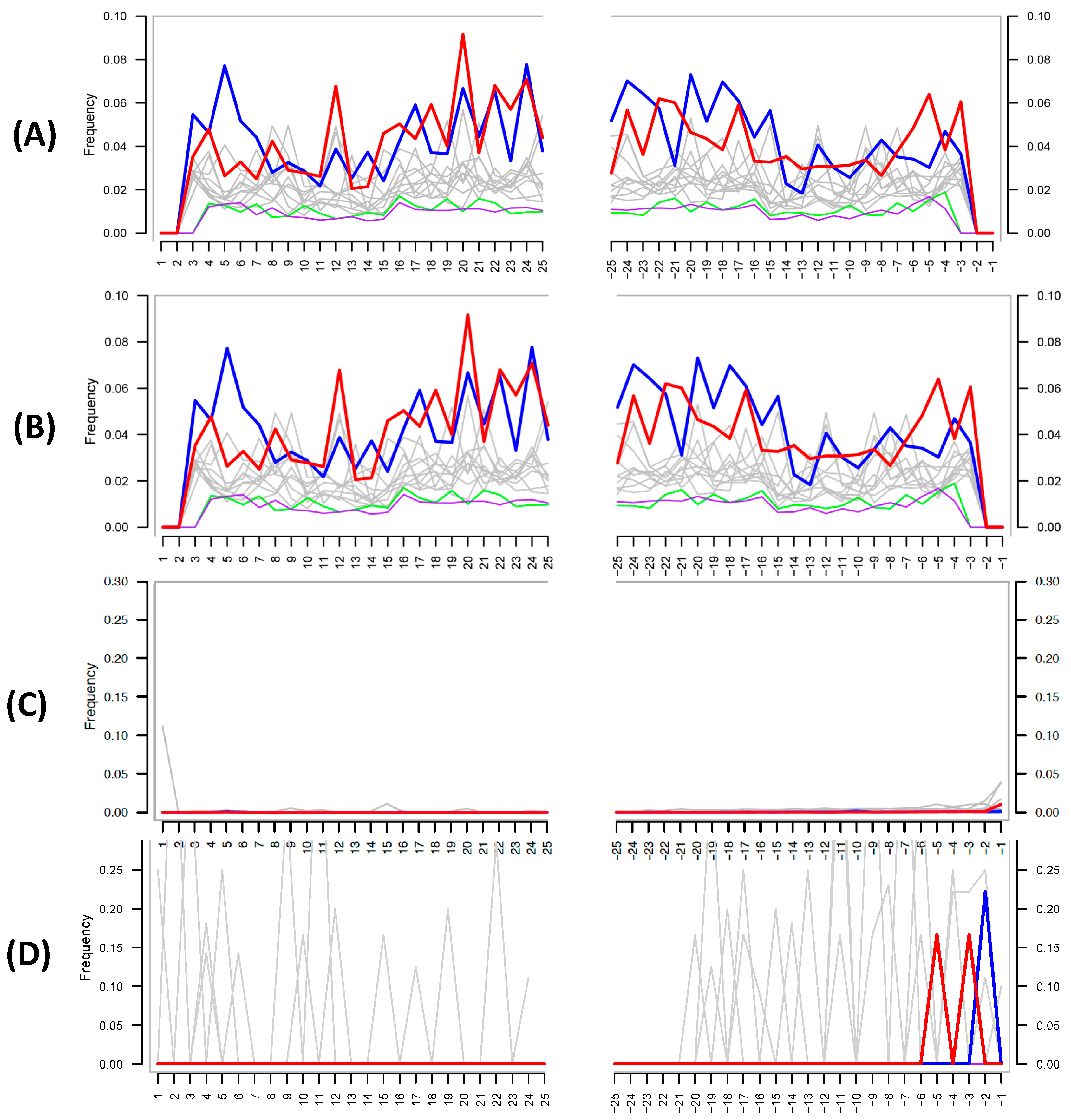
Proper Authentication of Ancient DNA Is Still Essential


{kind=link}
{kind=link}
Author Contributions
Conflicts of Interest
References
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Toranzos, G.A.; Marota, I.; Giuffra, V.; Cano, R.J. Gut Microbiome and Putative Resistome of Inca and Italian Nobility Mummies. Genes 2017, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, L.S.; Llamas, B.; Cooper, A. Reply to Santiago-Rodriguez et al.: Was luxS really isolated from 25- to 40-million-year-old bacteria? FEMS Microbiol. Lett. 2014, 353, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Cooper, A.; Weyrich, L.S. Reply to Santiago-Rodriguez et al.: Proper authentication of ancient DNA is essential. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Poinar, H.N. Ancient DNA: Do It Right or Not at All. Science 2000, 289, 1139. [Google Scholar] [CrossRef] [PubMed]
- Warinner, C.; Herbig, A.; Mann, A.; Yates, J.A.F.; Weiß, C.L.; Burbano, H.A.; Orlando, L.; Krause, J. A Robust Framework for Microbial Archaeology. Annu. Rev. Genom. Hum. Genet. 2017, 18, 321–356. [Google Scholar] [CrossRef] [PubMed]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. STAR Sci. Technol. Archaeol. Res. 2017, 3, 1–14. [Google Scholar] [CrossRef]
- Kim, D.; Hofstaedter, C.E.; Zhao, C.; Mattei, L.; Tanes, C.; Clarke, E.; Lauder, A.; Sherrill-Mix, S.; Chehoud, C.; Kelsen, J.; et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome 2017, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Rogelj, S.; Kieft, T.L. Sensitive, real-time PCR detects low-levels of contamination by Legionella pneumophila in commercial reagents. Mol. Cell. Probes 2006, 20, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Witt, N.; Rodger, G.; Vandesompele, J.; Benes, V.; Zumla, A.; Rook, G.A.; Huggett, J.F. An Assessment of Air As a Source of DNA Contamination Encountered When Performing PCR. J. Biomol. Tech. 2009, 20, 236–240. [Google Scholar] [PubMed]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. The Perils of Pathogen Discovery: Origin of a Novel Parvovirus-Like Hybrid Genome Traced to Nucleic Acid Extraction Spin Columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed]
- Lusk, R.W. Diverse and Widespread Contamination Evident in the Unmapped Depths of High Throughput Sequencing Data. PLoS ONE 2014, 9, e110808. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.I.; Bateman, A.C.; Bik, H.M.; Meadow, J.F. Microbiota of the indoor environment: a meta-analysis. Microbiome 2015, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Lauder, A.P.; Roche, A.M.; Sherrill-Mix, S.; Bailey, A.; Laughlin, A.L.; Bittinger, K.; Leite, R.; Elovitz, M.A.; Parry, S.; Bushman, F.D. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Perez-Muñoz, M.E.; Arrieta, M.-C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Key, F.M.; Posth, C.; Krause, J.; Herbig, A.; Bos, K.I. Mining Metagenomic Data Sets for Ancient DNA: Recommended Protocols for Authentication. Trends Genet. 2017, 33, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinforma. Oxf. Engl. 2013, 29, 1682–1684. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Fornaciari, G.; Luciani, S.; Dowd, S.E.; Toranzos, G.A.; Marota, I.; Cano, R.J. Taxonomic and predicted metabolic profiles of the human gut microbiome in pre-Columbian mummies. FEMS Microbiol. Ecol. 2016, 92, fiw182. [Google Scholar] [CrossRef] [PubMed]
- Ziesemer, K.A.; Mann, A.E.; Sankaranarayanan, K.; Schroeder, H.; Ozga, A.T.; Brandt, B.W.; Zaura, E.; Waters-Rist, A.; Hoogland, M.; Salazar-García, D.C.; et al. Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification. Sci. Rep. 2015, 5, 16498. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017, 544, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Valtueña, A.A.; Mittnik, A.; Key, F.M.; Haak, W.; Allmäe, R.; Belinskij, A.; Daubaras, M.; Feldman, M.; Jankauskas, R.; Janković, I.; et al. The Stone Age Plague and Its Persistence in Eurasia. Curr. Biol. 2017, 27, 3683–3691.e8. [Google Scholar] [CrossRef]
- Vågene, Å.J.; Herbig, A.; Campana, M.G.; García, N.M.R.; Warinner, C.; Sabin, S.; Spyrou, M.A.; Valtueña, A.A.; Huson, D.; Tuross, N.; et al. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico. Nat. Ecol. Evol. 2018, 2, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Herbig, A.; Maixner, F.; Bos, K.I.; Zink, A.; Krause, J.; Huson, D.H. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv 2016, 050559. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenhofer, R.; Weyrich, L.S. Proper Authentication of Ancient DNA Is Still Essential. Genes 2018, 9, 122. https://doi.org/10.3390/genes9030122
Eisenhofer R, Weyrich LS. Proper Authentication of Ancient DNA Is Still Essential. Genes. 2018; 9(3):122. https://doi.org/10.3390/genes9030122
Chicago/Turabian StyleEisenhofer, Raphael, and Laura S. Weyrich. 2018. "Proper Authentication of Ancient DNA Is Still Essential" Genes 9, no. 3: 122. https://doi.org/10.3390/genes9030122
APA StyleEisenhofer, R., & Weyrich, L. S. (2018). Proper Authentication of Ancient DNA Is Still Essential. Genes, 9(3), 122. https://doi.org/10.3390/genes9030122