Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology



Abstract
1. Introduction
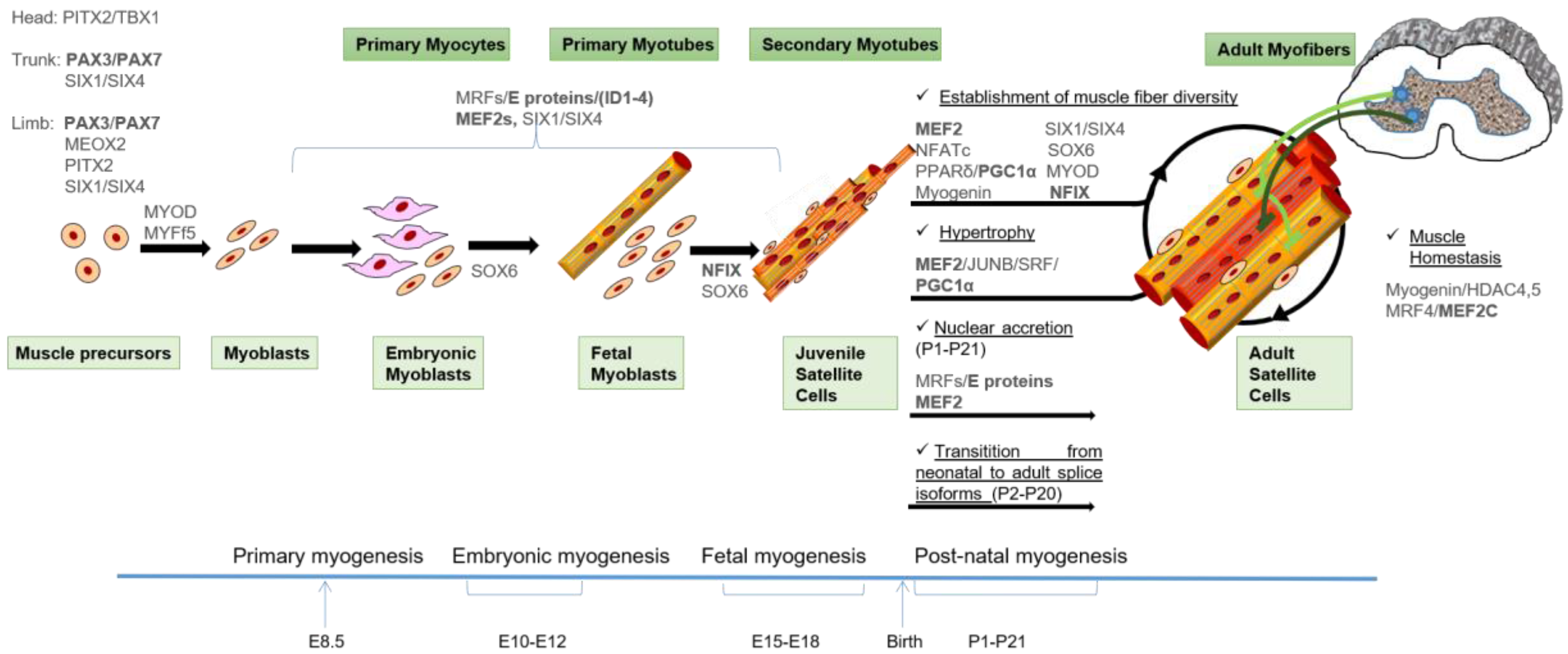
2. Myogenesis
2.1. Myogenesis During Embryonic Development
2.2. Post-Natal Myogenesis
3. Transcriptional Control of Skeletal Myogenesis
3.1. Families of Myogenic Transcription Factors
3.1.1. PAX3 and PAX7
3.1.2. SIX1 and SIX4
3.1.3. Myogenic Regulatory Factors
3.1.4. Repressors of MRFs
3.1.5. Co-Activators of MRFs
3.1.6. Myocyte Enhancer Factor-2 Proteins
3.2. Transcriptional Control of Specific Gene Expression Programs in Skeletal Muscle
3.2.1. Fetal Specific Gene Expression
3.2.2. Muscle Plasticity
3.2.3. Muscle Mass Homeostasis
4. Alternative Splicing in Skeletal Muscle
4.1. Alternative Splicing
4.2. Alternative Splicing in Skeletal Muscle Physiology
4.3. Aberrant Alternative Splicing in Skeletal Muscle Pathologies
5. Alternative Splicing of TFs in Skeletal Muscle Physiology and Muscle Diseases
5.1. Pax Family Genes
5.2. Myogenic Determination Factor Gene
5.3. Nuclear Factor 1/X Gene
5.4. Myocyte Enhancer Factor-2 Family Genes
5.5. Ubiquitous bHLH Proteins Class I
5.6. Inhibitor of DNA Binding (ID) Genes
6. Discussion
7. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Gurdon, J.B.; Bourillot, P.Y. Morphogen gradient interpretation. Nature 2001, 413, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.J. Developmental role of transcription factor isoforms generated by alternative splicing. Dev. Biol. 1995, 172, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Talavera, D.; Orozco, M.; de la Cruz, X. Alternative splicing of transcription factors’ genes: Beyond the increase of proteome diversity. Comp. Funct. Genom. 2009, 2009, 905894. [Google Scholar] [CrossRef] [PubMed]
- Taneri, B.; Snyder, B.; Novoradovsky, A.; Gaasterland, T. Alternative splicing of mouse transcription factors affects their DNA-binding domain architecture and is tissue specific. Genome Biol. 2004, 5, R75. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Ann. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Pistoni, M.; Ghigna, C.; Gabellini, D. Alternative splicing and muscular dystrophy. RNA Biol. 2010, 7, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [PubMed]
- Muntoni, F.; Wood, M.J. Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 2011, 10, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; la Rosa, P.; Sette, C. Faulty RNA splicing: Consequences and therapeutic opportunities in brain and muscle disorders. Hum. Genet. 2017, 136, 1215–1235. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J. Intern. Med. 2009, 266, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Comai, G.; Tajbakhsh, S. Molecular and cellular regulation of skeletal myogenesis. Curr. Top. Dev. Biol. 2014, 110, 1–73. [Google Scholar] [PubMed]
- Christ, B.; Ordahl, C.P. Early stages of chick somite development. Anat. Embryol. 1995, 191, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Magli, A.; Perlingeiro, R.R.C. Myogenic progenitor specification from pluripotent stem cells. Semin. Cell Dev. Biol. 2017, 72, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Biressi, S.; Molinaro, M.; Cossu, G. Cellular heterogeneity during vertebrate skeletal muscle development. Dev. Biol. 2007, 308, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M. Gene regulatory networks and cell lineages that underlie the formation of skeletal muscle. Proc. Natl. Acad. Sci. USA 2017, 114, 5830–5837. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Gautel, M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 2011, 12, 349–361. [Google Scholar]
- Duxson, M.J.; Usson, Y.; Harris, A.J. The origin of secondary myotubes in mammalian skeletal muscles: Ultrastructural studies. Development 1989, 107, 743–750. [Google Scholar] [PubMed]
- Vasyutina, E.; Birchmeier, C. The development of migrating muscle precursor cells. Anat. Embryol. 2006, 211, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Ontell, M.; Kozeka, K. The organogenesis of murine striated muscle: A cytoarchitectural study. Am. J. Anat. 1984, 171, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- White, R.B.; Bierinx, A.S.; Gnocchi, V.F.; Zammit, P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, F.; Rudnicki, M. Satellite and stem cells in muscle growth and repair. Development 2007, 134, 3953–3957. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Bottinelli, R.; Reggiani, C. Human skeletal muscle fibres: Molecular and functional diversity. Prog. Biophys. Mol. Biol. 2000, 73, 195–262. [Google Scholar] [CrossRef]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Pette, D.; Staron, R.S. Transitions of muscle fiber phenotypic profiles. Histochem. Cell Biol. 2001, 115, 359–372. [Google Scholar] [PubMed]
- Bassel-Duby, R.; Olson, E.N. Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.H.; Seale, P.; Rudnicki, M.A. Looking back to the embryo: Defining transcriptional networks in adult myogenesis. Nat. Rev. Genet. 2003, 4, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Lagha, M.; Brunelli, S.; Messina, G.; Cumano, A.; Kume, T.; Relaix, F.; Buckingham, M.E. Pax3:Foxc2 reciprocal repression in the somite modulates muscular versus vascular cell fate choice in multipotent progenitors. Dev. Cell 2009, 17, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Kassar-Duchossoy, L.; Giacone, E.; Gayraud-Morel, B.; Jory, A.; Gomes, D.; Tajbakhsh, S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 2005, 19, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005, 435, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Lagha, M.; Sato, T.; Bajard, L.; Daubas, P.; Esner, M.; Montarras, D.; Relaix, F.; Buckingham, M. Regulation of skeletal muscle stem cell behavior by Pax3 and Pax7. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Demignon, J.; Laclef, C.; Pujol, J.; Santolini, M.; Niro, C.; Lagha, M.; Rocancourt, D.; Buckingham, M.; Maire, P. Six homeoproteins directly activate Myod expression in the gene regulatory networks that control early myogenesis. PLoS Genet. 2013, 9, e1003425. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G.; Jerome-Majewska, L.A.; Papaioannou, V.E. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum. Mol. Genet. 2004, 13, 2829–2840. [Google Scholar] [CrossRef] [PubMed]
- Gage, P.J.; Suh, H.; Camper, S.A. Dosage requirement of Pitx2 for development of multiple organs. Development 1999, 126, 4643–4651. [Google Scholar] [PubMed]
- Kitamura, K.; Miura, H.; Miyagawa-Tomita, S.; Yanazawa, M.; Katoh-Fukui, Y.; Suzuki, R.; Ohuchi, H.; Suehiro, A.; Motegi, Y.; Nakahara, Y.; et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 1999, 126, 5749–5758. [Google Scholar] [PubMed]
- Zacharias, A.L.; Lewandoski, M.; Rudnicki, M.A.; Gage, P.J. Pitx2 is an upstream activator of extraocular myogenesis and survival. Dev. Biol. 2011, 349, 395–405. [Google Scholar] [CrossRef] [PubMed]
- L’Honore, A.; Ouimette, J.F.; Lavertu-Jolin, M.; Drouin, J. Pitx2 defines alternate pathways acting through MyoD during limb and somitic myogenesis. Development 2010, 137, 3847–3856. [Google Scholar] [CrossRef] [PubMed]
- Buchberger, A.; Freitag, D.; Arnold, H.H. A homeo-paired domain-binding motif directs Myf5 expression in progenitor cells of limb muscle. Development 2007, 134, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Giordani, J.; Bajard, L.; Demignon, J.; Daubas, P.; Buckingham, M.; Maire, P. Six proteins regulate the activation of Myf5 expression in embryonic mouse limbs. Proc. Natl. Acad. Sci. USA 2007, 104, 11310–11315. [Google Scholar] [CrossRef] [PubMed]
- Grifone, R.; Laclef, C.; Spitz, F.; Lopez, S.; Demignon, J.; Guidotti, J.E.; Kawakami, K.; Xu, P.X.; Kelly, R.; Petrof, B.J.; et al. Six1 and Eya1 expression can reprogram adult muscle from the slow-twitch phenotype into the fast-twitch phenotype. Mol. Cell. Biol. 2004, 24, 6253–6267. [Google Scholar] [CrossRef] [PubMed]
- Grifone, R.; Demignon, J.; Houbron, C.; Souil, E.; Niro, C.; Seller, M.J.; Hamard, G.; Maire, P. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 2005, 132, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Laclef, C.; Hamard, G.; Demignon, J.; Souil, E.; Houbron, C.; Maire, P. Altered myogenesis in Six1-deficient mice. Development 2003, 130, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Lassar, A.B.; Paterson, B.M.; Weintraub, H. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell 1986, 47, 649–656. [Google Scholar] [CrossRef]
- Berkes, C.A.; Bergstrom, D.A.; Penn, B.H.; Seaver, K.J.; Knoepfler, P.S.; Tapscott, S.J. Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol. Cell 2004, 14, 465–477. [Google Scholar] [CrossRef]
- Gerber, A.N.; Klesert, T.R.; Bergstrom, D.A.; Tapscott, S.J. Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: A mechanism for lineage determination in myogenesis. Genes Dev. 1997, 11, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Kassar-Duchossoy, L.; Gayraud-Morel, B.; Gomes, D.; Rocancourt, D.; Buckingham, M.; Shinin, V.; Tajbakhsh, S. Mrf4 determines skeletal muscle identity in Myf5:Myod double-mutant mice. Nature 2004, 431, 466–471. [Google Scholar] [CrossRef] [PubMed]
- De la Serna, I.L.; Carlson, K.A.; Imbalzano, A.N. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat. Genet. 2001, 27, 187–190. [Google Scholar] [CrossRef] [PubMed]
- De la Serna, I.L.; Ohkawa, Y.; Berkes, C.A.; Bergstrom, D.A.; Dacwag, C.S.; Tapscott, S.J.; Imbalzano, A.N. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol. Cell. Biol. 2005, 25, 3997–4009. [Google Scholar] [CrossRef] [PubMed]
- Forcales, S.V.; Albini, S.; Giordani, L.; Malecova, B.; Cignolo, L.; Chernov, A.; Coutinho, P.; Saccone, V.; Consalvi, S.; Williams, R.; et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2012, 31, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.L.; Sartorelli, V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell. Physiol. 2000, 185, 155–173. [Google Scholar] [CrossRef]
- Berkes, C.A.; Tapscott, S.J. MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 2005, 16, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Tapscott, S.J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 2005, 132, 2685–2695. [Google Scholar] [CrossRef] [PubMed]
- Penn, B.H.; Bergstrom, D.A.; Dilworth, F.J.; Bengal, E.; Tapscott, S.J. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 2004, 18, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Biesiada, E.; Hamamori, Y.; Kedes, L.; Sartorelli, V. Myogenic basic helix-loop-helix proteins and Sp1 interact as components of a multiprotein transcriptional complex required for activity of the human cardiac alpha-actin promoter. Mol. Cell. Biol. 1999, 19, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.G.; Cheng, T.C.; Cserjesi, P.; Chakraborty, T.; Olson, E.N. Analysis of the myogenin promoter reveals an indirect pathway for positive autoregulation mediated by the muscle-specific enhancer factor MEF-2. Mol. Cell. Biol. 1992, 12, 3665–3677. [Google Scholar] [CrossRef] [PubMed]
- Amacher, S.L.; Buskin, J.N.; Hauschka, S.D. Multiple regulatory elements contribute differentially to muscle creatine kinase enhancer activity in skeletal and cardiac muscle. Mol. Cell. Biol. 1993, 13, 2753–2764. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Olson, E.N. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. USA 1996, 93, 9366–9373. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.V.; Hughes, S.M. Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 2017, 72, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Ranganayakulu, G.; Zhao, B.; Dokidis, A.; Molkentin, J.D.; Olson, E.N.; Schulz, R.A. A series of mutations in the D-MEF2 transcription factor reveal multiple functions in larval and adult myogenesis in Drosophila. Dev. Biol. 1995, 171, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Bour, B.A.; O’Brien, M.A.; Lockwood, W.L.; Goldstein, E.S.; Bodmer, R.; Taghert, P.H.; Abmayr, S.M.; Nguyen, H.T. Drosophila MEF2, a transcription factor that is essential for myogenesis. Genes Dev. 1995, 9, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Lilly, B.; Zhao, B.; Ranganayakulu, G.; Paterson, B.M.; Schulz, R.A.; Olson, E.N. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science 1995, 267, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.V.; Nadal-Ginard, B. Early expression of the different isoforms of the myocyte enhancer factor-2 (MEF2) protein in myogenic as well as non-myogenic cell lineages during mouse embryogenesis. Mech. Dev. 1996, 57, 103–112. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Arnold, M.A.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol. Cell. Biol. 2007, 27, 8143–8151. [Google Scholar] [CrossRef] [PubMed]
- Hinits, Y.; Hughes, S.M. Mef2s are required for thick filament formation in nascent muscle fibres. Development 2007, 134, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Molinari, S.; Melchionna, R.; Angelis, M.G.C.; Battini, R.; de Angelis, L.; Kelly, R.; Cossu, G. Absence of MEF2 binding to the A/T-rich element in the muscle creatine kinase (MCK) enhancer correlates with lack of early expression of the MCK gene in embryonic mammalian muscle. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 23–34. [Google Scholar]
- Messina, G.; Biressi, S.; Monteverde, S.; Magli, A.; Cassano, M.; Perani, L.; Roncaglia, E.; Tagliafico, E.; Starnes, L.; Campbell, C.E.; et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 2010, 140, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Baruffaldi, F.; Montarras, D.; Basile, V.; de Feo, L.; Badodi, S.; Ganassi, M.; Battini, R.; Nicoletti, C.; Imbriano, C.; Musaro, A.; et al. Dynamic phosphorylation of the myocyte enhancer factor 2Calpha1 splice variant promotes skeletal muscle regeneration and hypertrophy. Stem Cells 2017, 35, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Moretti, I.; Ciciliot, S.; Dyar, K.A.; Abraham, R.; Murgia, M.; Agatea, L.; Akimoto, T.; Bicciato, S.; Forcato, M.; Pierre, P.; et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat. Commun. 2016, 7, 12397. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Hu, J.; Barnes, R.M.; Heidt, A.B.; Cornelissen, I.; Black, B.L. Myocyte enhancer factor 2C function in skeletal muscle is required for normal growth and glucose metabolism in mice. Skelet. Muscle 2015, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 2001, 11, 497–504. [Google Scholar] [CrossRef]
- Bertos, N.R.; Wang, A.H.; Yang, X.J. Class II histone deacetylases: Structure, function, and regulation. Biochem. Cell Biol. 2001, 79, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Maroli, G.; Cermenati, S.; Monteverde, S.; Ferrante, A.; Rossi, G.; Cossu, G.; Beltrame, M.; Messina, G. Nfix Induces a switch in Sox6 transcriptional activity to regulate MyHC-I expression in fetal muscle. Cell Rep. 2016, 17, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Yeh, M.; Liu, A. Sox6 is required for normal fiber type differentiation of fetal skeletal muscle in mice. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 2062–2076. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.F.; Demignon, J.; Sakakibara, I.; Pujol, J.; Favier, M.; Strochlic, L.; le Grand, F.; Sgarioto, N.; Guernec, A.; Schmitt, A.; et al. Genesis of muscle fiber-type diversity during mouse embryogenesis relies on Six1 and Six4 gene expression. Dev. Biol. 2011, 359, 303–320. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. MEF2: A calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 2002, 27, 40–47. [Google Scholar] [CrossRef]
- McCullagh, K.J.; Calabria, E.; Pallafacchina, G.; Ciciliot, S.; Serrano, A.L.; Argentini, C.; Kalhovde, J.M.; Lomo, T.; Schiaffino, S. NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl. Acad. Sci. USA 2004, 101, 10590–10595. [Google Scholar] [CrossRef] [PubMed]
- Calabria, E.; Ciciliot, S.; Moretti, I.; Garcia, M.; Picard, A.; Dyar, K.A.; Pallafacchina, G.; Tothova, J.; Schiaffino, S.; Murgia, M. NFAT isoforms control activity-dependent muscle fiber type specification. Proc. Natl. Acad. Sci. USA 2009, 106, 13335–13340. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Rothermel, B.; Kanatous, S.; Rosenberg, P.; Naya, F.J.; Shelton, J.M.; Hutcheson, K.A.; DiMaio, J.M.; Olson, E.N.; Bassel-Duby, R.; et al. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001, 20, 6414–6423. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Lunde, I.G.; Ekmark, M.; Rana, Z.A.; Buonanno, A.; Gundersen, K. PPARdelta expression is influenced by muscle activity and induces slow muscle properties in adult rat muscles after somatic gene transfer. J. Physiol. 2007, 582, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef] [PubMed]
- Ekmark, M.; Rana, Z.A.; Stewart, G.; Hardie, D.G.; Gundersen, K. De-phosphorylation of MyoD is linking nerve-evoked activity to fast myosin heavy chain expression in rodent adult skeletal muscle. J. Physiol. 2007, 584, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.M.; Chi, M.M.; Lowry, O.H.; Gundersen, K. Myogenin induces a shift of enzyme activity from glycolytic to oxidative metabolism in muscles of transgenic mice. J. Cell Biol. 1999, 145, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Blaauw, B.; Canato, M.; Agatea, L.; Toniolo, L.; Mammucari, C.; Masiero, E.; Abraham, R.; Sandri, M.; Schiaffino, S.; Reggiani, C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3896–3905. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Signaling in muscle atrophy and hypertrophy. Physiology 2008, 23, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; del Piccolo, P.; Burden, S.J.; di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Czubryt, M.P.; McAnally, J.; Bassel-Duby, R.; Richardson, J.A.; Wiebel, F.F.; Nordheim, A.; Olson, E.N. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Parlakian, A.; Tuil, D.; Hamard, G.; Tavernier, G.; Hentzen, D.; Concordet, J.P.; Paulin, D.; Li, Z.; Daegelen, D. Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Mol. Cell. Biol. 2004, 24, 5281–5289. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; Milan, G.; Masiero, E.; Carnio, S.; Lee, D.; Lanfranchi, G.; Goldberg, A.L.; Sandri, M. JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J. Cell Biol. 2010, 191, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, X.; Qian, Y.; LaDuca, J.P.; Maquat, L.E. At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: A possible link between nuclear splicing and cytoplasmic translation. Mol. Cell. Biol. 1998, 18, 5272–5283. [Google Scholar] [CrossRef] [PubMed]
- Thermann, R.; Neu-Yilik, G.; Deters, A.; Frede, U.; Wehr, K.; Hagemeier, C.; Hentze, M.W.; Kulozik, A.E. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998, 17, 3484–3494. [Google Scholar] [CrossRef] [PubMed]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Schor, I.E.; Allo, M.; Dujardin, G.; Petrillo, E.; Munoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Warren, S.L. A functional interaction between the carboxy-terminal domain of RNA polymerase II and pre-mRNA splicing. J. Cell Biol. 1997, 136, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Yao, X.; Lin, Q.J.; Yin, J.W.; Liang, Y.; Heiner, M.; Tian, B.; Hui, J.; Wang, G. Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol. Cell 2012, 45, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Ma, X.; Meruvu, S.; Hugendubler, L.; Mueller, E. The adipogenic transcriptional cofactor ZNF638 interacts with splicing regulators and influences alternative splicing. J. Lipid Res. 2014, 55, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Dowhan, D.H.; Kang, Y.K.; Larkin, K.; Lee, J.W.; Berget, S.M.; O’Malley, B.W. Differential recruitment of nuclear receptor coactivators may determine alternative RNA splice site choice in target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2270–2274. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Llorian, M.; Smith, C.W. Decoding muscle alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.S.; Wang, E.T.; Vu, A.; David, M.P.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010, 38, 7651–7664. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.H.; Charlet, B.N.; Poulos, M.G.; Singh, G.; Swanson, M.S.; Cooper, T.A. Muscleblind proteins regulate alternative splicing. EMBO J. 2004, 23, 3103–3112. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Oma, Y.; Onishi, H.; Nezu, Y.; Sasagawa, N.; Nukina, N.; Ishiura, S. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res. 2009, 37, 6477–6490. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Vicente, M.; Monferrer, L.; Artero, R. The Muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation 2006, 74, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Spellman, R.; Smith, C.W. Novel modes of splicing repression by PTB. Trends Biochem. Sci. 2006, 31, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hung, L.H.; Licht, T.; Kostin, S.; Looso, M.; Khrameeva, E.; Bindereif, A.; Schneider, A.; Braun, T. RBM24 is a major regulator of muscle-specific alternative splicing. Dev. Cell 2014, 31, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Tarn, W.Y. RBM4 down-regulates PTB and antagonizes its activity in muscle cell-specific alternative splicing. J. Cell Biol. 2011, 193, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Cooper, T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Kearney, D.L.; de Biasi, M.; Taffet, G.; Cooper, T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Investig. 2007, 117, 2802–2811. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Cline, M.S.; Osborne, R.J.; Tuttle, D.L.; Clark, T.A.; Donohue, J.P.; Hall, M.P.; Shiue, L.; Swanson, M.S.; Thornton, C.A.; Ares, M., Jr. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010, 17, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Osborne, R.J.; Lin, X.; Welle, S.; Sobczak, K.; O’Rourke, J.R.; Swanson, M.S.; Thornton, C.A. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 2009, 18, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.F.; Gasnier, E.; Furling, D. Gain of RNA function in pathological cases: Focus on myotonic dystrophy. Biochimie 2011, 93, 2006–2012. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Sznajder, L.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Sancisi, V.; Germinario, E.; Esposito, A.; Morini, E.; Peron, S.; Moggio, M.; Tomelleri, G.; Danieli-Betto, D.; Tupler, R. Altered Tnnt3 characterizes selective weakness of fast fibers in mice overexpressing FSHD region gene 1 (FRG1). Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R124–R137. [Google Scholar] [CrossRef] [PubMed]
- Gabellini, D.; D’Antona, G.; Moggio, M.; Prelle, A.; Zecca, C.; Adami, R.; Angeletti, B.; Ciscato, P.; Pellegrino, M.A.; Bottinelli, R.; et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature 2006, 439, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Pistoni, M.; Shiue, L.; Cline, M.S.; Bortolanza, S.; Neguembor, M.V.; Xynos, A.; Ares, M., Jr.; Gabellini, D. Rbfox1 downregulation and altered calpain 3 splicing by FRG1 in a mouse model of Facioscapulohumeral muscular dystrophy (FSHD). PLoS Genet. 2013, 9, e1003186. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Differentially expressed alternatively spliced genes in skeletal muscle from cancer patients with cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Orengo, J.P.; Ward, A.J.; Cooper, T.A. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol. 2011, 69, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, A.; Manni, I.; Fuschi, P.; Mantovani, R.; Guadagni, F.; Sacchi, A.; Piaggio, G. Requirement for down-regulation of the CCAAT-binding activity of the NF-Y transcription factor during skeletal muscle differentiation. Mol. Biol. Cell 2003, 14, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Basile, V.; Baruffaldi, F.; Dolfini, D.; Belluti, S.; Benatti, P.; Ricci, L.; Artusi, V.; Tagliafico, E.; Mantovani, R.; Molinari, S.; Imbriano, C. NF-YA splice variants have different roles on muscle differentiation. Biochim. Biophys. Acta 2016, 1859, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.A. PAX proteins and fables of their reconstruction. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, D.; Jimenez, G.; Heavey, B.; Busslinger, M. Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J. 2000, 19, 2292–2303. [Google Scholar] [CrossRef] [PubMed]
- Lamey, T.M.; Koenders, A.; Ziman, M. Pax genes in myogenesis: Alternate transcripts add complexity. Histol. Histopathol. 2004, 19, 1289–1300. [Google Scholar] [PubMed]
- Ziman, M.R.; Fletcher, S.; Kay, P.H. Alternate Pax7 transcripts are expressed specifically in skeletal muscle, brain and other organs of adult mice. Int. J. Biochem. Cell Biol. 1997, 29, 1029–1036. [Google Scholar] [CrossRef]
- Vogan, K.J.; Underhill, D.A.; Gros, P. An alternative splicing event in the Pax-3 paired domain identifies the linker region as a key determinant of paired domain DNA-binding activity. Mol. Cell. Biol. 1996, 16, 6677–6686. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Lawrence, E.J.; Strzelecki, D.; Rajput, P.; Xia, S.J.; Gottesman, D.M.; Barr, F.G. Co-expression of alternatively spliced forms of PAX3, PAX7, PAX3-FKHR and PAX7-FKHR with distinct DNA binding and transactivation properties in rhabdomyosarcoma. Int. J. Cancer 2005, 115, 85–92. [Google Scholar] [CrossRef] [PubMed]
- White, R.B.; Ziman, M.R. Genome-wide discovery of Pax7 target genes during development. Physiol. Genom. 2008, 33, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.C.; Saetre, B.O.; Havik, B.; Ellingsen, S.; Fjose, A. The zebrafish Pax3 and Pax7 homologues are highly conserved, encode multiple isoforms and show dynamic segment-like expression in the developing brain. Mech. Dev. 1998, 70, 49–63. [Google Scholar] [CrossRef]
- Vogan, K.J.; Gros, P. The C-terminal subdomain makes an important contribution to the DNA binding activity of the Pax-3 paired domain. J. Biol. Chem. 1997, 272, 28289–28295. [Google Scholar] [CrossRef] [PubMed]
- Czerny, T.; Schaffner, G.; Busslinger, M. DNA sequence recognition by Pax proteins: Bipartite structure of the paired domain and its binding site. Genes Dev. 1993, 7, 2048–2061. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Fitzgerald, J.C.; Ginsberg, J.P.; Vanella, M.L.; Davis, R.J.; Bennicelli, J.L. Predominant expression of alternative PAX3 and PAX7 forms in myogenic and neural tumor cell lines. Cancer Res. 1999, 59, 5443–5448. [Google Scholar] [PubMed]
- Pritchard, C.; Grosveld, G.; Hollenbach, A.D. Alternative splicing of Pax3 produces a transcriptionally inactive protein. Gene 2003, 305, 61–69. [Google Scholar] [CrossRef]
- Charytonowicz, E.; Matushansky, I.; Castillo-Martin, M.; Hricik, T.; Cordon-Cardo, C.; Ziman, M. Alternate PAX3 and PAX7 C-terminal isoforms in myogenic differentiation and sarcomagenesis. Clin. Transl. Oncol. 2011, 13, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kumar, S.; Slevin, M.; Kumar, P. Functional analysis of alternative isoforms of the transcription factor PAX3 in melanocytes in vitro. Cancer Res. 2006, 66, 8574–8580. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.D.; Barber, M.C.; Cloutier, T.E.; Friedman, T.B. PAX3 gene structure, alternative splicing and evolution. Gene 1999, 237, 311–319. [Google Scholar] [CrossRef]
- Vorobyov, E.; Horst, J. Expression of two protein isoforms of PAX7 is controlled by competing cleavage-polyadenylation and splicing. Gene 2004, 342, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Hu, X.; Li, N. Identification and expression profile of a novel alternative splicing of Pax7 in chick skeletal muscle. Poult. Sci. 2008, 87, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.M.; Kinghorn, J.R.; Johnston, I.A. Differential regulation of multiple alternatively spliced transcripts of MyoD. Gene 2007, 391, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Meedel, T.H.; Farmer, S.C.; Lee, J.J. The single MyoD family gene of Ciona intestinalis encodes two differentially expressed proteins: Implications for the evolution of chordate muscle gene regulation. Development 1997, 124, 1711–1721. [Google Scholar] [PubMed]
- Rhodes, S.J.; Konieczny, S.F. Identification of MRF4: A new member of the muscle regulatory factor gene family. Genes Dev. 1989, 3, 2050–2061. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.J.; Chakraborty, T.; Martin, J.; Zhou, J.M.; Olson, E.N. The basic region of myogenin cooperates with two transcription activation domains to induce muscle-specific transcription. Mol. Cell. Biol. 1992, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Kudou, K.; Komatsu, T.; Nogami, J.; Maehara, K.; Harada, A.; Saeki, H.; Oki, E.; Maehara, Y.; Ohkawa, Y. The requirement of Mettl3-promoted MyoD mRNA maintenance in proliferative myoblasts for skeletal muscle differentiation. Open Biol. 2017, 7, 170119. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5’ UTR m(6)A Promotes Cap-Independent translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Grunder, A.; Qian, F.; Ebel, T.T.; Mincheva, A.; Lichter, P.; Kruse, U.; Sippel, A.E. Genomic organization, splice products and mouse chromosomal localization of genes for transcription factor Nuclear Factor One. Gene 2003, 304, 171–181. [Google Scholar] [CrossRef]
- Perfetti, A.; Greco, S.; Fasanaro, P.; Bugiardini, E.; Cardani, R.; Garcia-Manteiga, J.M.; Riba, M.; Cittaro, D.; Stupka, E.; Meola, G.; Martelli, F. Genome wide identification of aberrant alternative splicing events in myotonic dystrophy type 2. PLoS ONE 2014, 9, e93983. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Marin-Reina, P.; Sanchis-Calvo, A.; Perez-Aytes, A.; Oltra, S.; Rosello, M.; Mayo, S.; Monfort, S.; Pantoja, J.; Orellana, C. Novel mutations of NFIX gene causing Marshall-Smith syndrome or Sotos-like syndrome: One gene, two phenotypes. Pediatr. Res. 2015, 78, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Ganassi, M.; Badodi, S.; Polacchini, A.; Baruffaldi, F.; Battini, R.; Hughes, S.M.; Hinits, Y.; Molinari, S. Distinct functions of alternatively spliced isoforms encoded by zebrafish mef2ca and mef2cb. Biochim. Biophys. Acta 2014, 1839, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Faralli, H.; Yao, Z.; Rakopoulos, P.; Palii, C.; Cao, Y.; Singh, K.; Liu, Q.C.; Chu, A.; Aziz, A.; et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 2013, 27, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhu, B.; Davie, J. Alternative splicing of MEF2C pre-mRNA controls its activity in normal myogenesis and promotes tumorigenicity in Rhabdomyosarcoma cells. J. Biol. Chem. 2015, 290, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ren, S.; Lee, J.H.; Qiu, J.; Chapski, D.J.; Rau, C.D.; Zhou, Y.; Abdellatif, M.; Nakano, A.; Vondriska, T.M.; et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J. Clin. Investig. 2016, 126, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Badodi, S.; Baruffaldi, F.; Ganassi, M.; Battini, R.; Molinari, S. Phosphorylation-dependent degradation of MEF2C contributes to regulate G2/M transition. Cell Cycle 2015, 14, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ramachandran, B.; Gulick, T. Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J. Biol. Chem. 2005, 280, 28749–28760. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Gulick, T. Phosphorylation and alternative pre-mRNA splicing converge to regulate myocyte enhancer factor 2C activity. Mol. Cell. Biol. 2004, 24, 8264–8275. [Google Scholar] [CrossRef] [PubMed]
- Genikhovich, G.; Technau, U. Complex functions of Mef2 splice variants in the differentiation of endoderm and of a neuronal cell type in a sea anemone. Development 2011, 138, 4911–4919. [Google Scholar] [CrossRef] [PubMed]
- Della Gaspera, B.; Armand, A.S.; Sequeira, I.; Lecolle, S.; Gallien, C.L.; Charbonnier, F.; Chanoine, C. The Xenopus MEF2 gene family: Evidence of a role for XMEF2C in larval tendon development. Dev. Biol. 2009, 328, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, D.; Beatty, K.E.; Taylor, M.V. Different levels, but not different isoforms, of the Drosophila transcription factor DMEF2 affect distinct aspects of muscle differentiation. Dev. Biol. 1999, 215, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Spring, J.; Yanze, N.; Josch, C.; Middel, A.M.; Winninger, B.; Schmid, V. Conservation of Brachyury, Mef2, and Snail in the myogenic lineage of jellyfish: A connection to the mesoderm of bilateria. Dev. Biol. 2002, 244, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Bachinski, L.L.; Sirito, M.; Bohme, M.; Baggerly, K.A.; Udd, B.; Krahe, R. Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve 2010, 42, 856–863. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.C.; Cardoso, M.C.; Yu, Y.T.; Andres, V.; Leifer, D.; Krainc, D.; Lipton, S.A.; Nadal-Ginard, B. hMEF2C gene encodes skeletal muscle- and brain-specific transcription factors. Mol. Cell. Biol. 1993, 13, 2564–2577. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Cao, Y.; Witwicka, H.E.; Tom, S.; Tapscott, S.J.; Wang, E.H. RNA-binding protein Muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) {beta}-exon splicing. J. Biol. Chem. 2010, 285, 33779–33787. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.; Tremblay, A.M.; Xiao, L.; Yang, Q.; Ma, K.; Nie, J.; Mao, Z.; Wu, Z.; Giguere, V.; Yang, X.J. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J. Biol. Chem. 2006, 281, 4423–4433. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Gocke, C.B.; Yu, H. Phosphorylation-facilitated sumoylation of MEF2C negatively regulates its transcriptional activity. BMC Biochem. 2006, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Shalizi, A.; Gaudilliere, B.; Yuan, Z.; Stegmuller, J.; Shirogane, T.; Ge, Q.; Tan, Y.; Schulman, B.; Harper, J.W.; Bonni, A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 2006, 311, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sternsdorf, T.; Bolger, T.A.; Evans, R.M.; Yao, T.P. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol. Cell. Biol. 2005, 25, 8456–8464. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, C.; Barthel, K.K.; Liu, X. SUMO-1 modification of MEF2A regulates its transcriptional activity. J. Cell. Mol. Med. 2006, 10, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007, 14, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.F.; Miano, J.M.; Hustad, C.M.; Copeland, N.G.; Jenkins, N.A.; Olson, E.N. A Mef2 gene that generates a muscle-specific isoform via alternative mRNA splicing. Mol. Cell. Biol. 1994, 14, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Magli, A.; Angelelli, C.; Ganassi, M.; Baruffaldi, F.; Matafora, V.; Battini, R.; Bachi, A.; Messina, G.; Rustighi, A.; del Sal, G.; et al. Proline isomerase Pin1 represses terminal differentiation and myocyte enhancer factor 2C function in skeletal muscle cells. J. Biol. Chem. 2010, 285, 34518–34527. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Gagliostro, E.; Clocchiatti, A.; Brancolini, C. The control operated by the cell cycle machinery on MEF2 stability contributes to the downregulation of CDKN1A and entry into S phase. Mol. Cell. Biol. 2015, 35, 1633–1647. [Google Scholar] [CrossRef] [PubMed]
- Merlino, G.; Helman, L.J. Rhabdomyosarcoma—Working out the pathways. Oncogene 1999, 18, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Boutz, P.L.; Stoilov, P.; Li, Q.; Lin, C.H.; Chawla, G.; Ostrow, K.; Shiue, L.; Ares, M., Jr.; Black, D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007, 21, 1636–1652. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Xia, Z.; Bland, C.S.; Kalsotra, A.; Scavuzzo, M.A.; Curk, T.; Ule, J.; Li, W.; Cooper, T.A. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol. Cell 2014, 55, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Runfola, V.; Sebastian, S.; Dilworth, F.J.; Gabellini, D. Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J. Cell Sci. 2015, 128, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Watada, H.; Kajimoto, Y.; Umayahara, Y.; Matsuoka, T.; Morishima, T.; Yamasaki, Y.; Kawamori, R.; Kamada, T. Ubiquitous, but variable, expression of two alternatively spliced mRNAs encoding mouse homologues of transcription factors E47 and E12. Gene 1995, 153, 255–259. [Google Scholar] [CrossRef]
- Murre, C.; McCaw, P.S.; Vaessin, H.; Caudy, M.; Jan, L.Y.; Jan, Y.N.; Cabrera, C.V.; Buskin, J.N.; Hauschka, S.D.; Lassar, A.B.; et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 1989, 58, 537–544. [Google Scholar] [CrossRef]
- Yang, Z.; MacQuarrie, K.L.; Analau, E.; Tyler, A.E.; Dilworth, F.J.; Cao, Y.; Diede, S.J.; Tapscott, S.J. MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes Dev. 2009, 23, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.S.; Simmons, D.M.; Swanson, L.W.; Rosenfeld, M.G. Tissue-specific RNA splicing generates an ankyrin-like domain that affects the dimerization and DNA-binding properties of a bHLH protein. Genes Dev. 1993, 7, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.H.; Perry, R.L.; Fauteux, M.C.; Berkes, C.A.; Rudnicki, M.A. MyoD synergizes with the E-protein HEB beta to induce myogenic differentiation. Mol. Cell. Biol. 2006, 26, 5771–5783. [Google Scholar] [CrossRef] [PubMed]
- Corneliussen, B.; Thornell, A.; Hallberg, B.; Grundstrom, T. Helix-loop-helix transcriptional activators bind to a sequence in glucocorticoid response elements of retrovirus enhancers. J. Virol. 1991, 65, 6084–6093. [Google Scholar] [PubMed]
- Sepp, M.; Kannike, K.; Eesmaa, A.; Urb, M.; Timmusk, T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5’ exon usage and splicing. PLoS ONE 2011, 6, e22138. [Google Scholar] [CrossRef] [PubMed]
- Benezra, R.; Davis, R.L.; Lockshon, D.; Turner, D.L.; Weintraub, H. The protein Id: A negative regulator of helix-loop-helix DNA binding proteins. Cell 1990, 61, 49–59. [Google Scholar] [CrossRef]
- Roschger, C.; Cabrele, C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun. Signal. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Jen, Y.; Weintraub, H.; Benezra, R. Overexpression of Id protein inhibits the muscle differentiation program: In vivo association of Id with E2A proteins. Genes Dev. 1992, 6, 1466–1479. [Google Scholar] [CrossRef] [PubMed]
- Nehlin, J.O.; Hara, E.; Kuo, W.L.; Collins, C.; Campisi, J. Genomic organization, sequence, and chromosomal localization of the human helix-loop-helix Id1 gene. Biochem. Biophys. Res. Commun. 1997, 231, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Deed, R.W.; Jasiok, M.; Norton, J.D. Attenuated function of a variant form of the helix-loop-helix protein, Id-3, generated by an alternative splicing mechanism. FEBS Lett. 1996, 393, 113–116. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Lancet, D.; Yanai, I. Alternative splicing and gene duplication are inversely correlated evolutionary mechanisms. Nat. Genet. 2005, 37, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wang, J.; Yu, J.; Huang, X.; Gu, X. Evolution of alternative splicing after gene duplication. Genome Res. 2006, 16, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Robinson-Rechavi, M. Age-dependent gain of alternative splice forms and biased duplication explain the relation between splicing and duplication. Genome Res. 2011, 21, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Cannavino, J.; Brocca, L.; Sandri, M.; Bottinelli, R.; Pellegrino, M.A. PGC1-alpha over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J. Physiol. 2014, 592, 4575–4589. [Google Scholar] [CrossRef] [PubMed]
- Correia, J.C.; Ferreira, D.M.; Ruas, J.L. Intercellular: Local and systemic actions of skeletal muscle PGC-1s. Trends Endocrinol. Metab. 2015, 26, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T. PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life 2009, 61, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1alpha isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol. Cell 2000, 6, 307–316. [Google Scholar] [CrossRef]
- Brinegar, A.E.; Cooper, T.A. Roles for RNA-binding proteins in development and disease. Brain Res. 2016, 1647, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bachinski, L.L.; Baggerly, K.A.; Neubauer, V.L.; Nixon, T.J.; Raheem, O.; Sirito, M.; Unruh, A.K.; Zhang, J.; Nagarajan, L.; Timchenko, L.T.; et al. Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul. Disord. 2014, 24, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Ackall, F.Y.; Hur, J.; Sharma, K.; Paulson, H.L.; Dowling, J.J. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis. Models Mech. 2014, 7, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Ebralidze, A.; Wang, Y.; Petkova, V.; Ebralidse, K.; Junghans, R.P. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science 2004, 303, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Tupler, R.; Perini, G.; Pellegrino, M.A.; Green, M.R. Profound misregulation of muscle-specific gene expression in facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 12650–12654. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Thornton, Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, N. Modulation of aberrant splicing in human RNA diseases by chemical compounds. Hum. Genet. 2017, 136, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from Myotonic Dystrophy 1 patients. Mol. Ther. Nucleic Acids 2017, 9, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]

| Type of alternative splicing | TFs | Effects on TF |
| Exon skipping/retention  | NFIX1/2/3 | NFIX2: regulator of fetal muscle genes |
| HEBα/β | HEBα: higher DNA binding and transcriptional activity
HEBβ: synergises with MyoD in early differentiation | |
| PAX3Q+/− | PAX3Q-: stronger binding affinity and higher transcriptional activity | |
| PAX7Q+/− | NA | |
| PAX7GL+/− | PAX7GL-: increased binding affinity | |
| PAX7-2 | PAX7-2: reduced transcriptional activity | |
| E12/47
E2A-2/5 | E2A-2/5: dominant negative activity | |
| MEF2A/B/C β | MEF2A/B/C β: higher transcriptional activity | |
| TCF4 A-R | TCF4 AD1-containing isoforms: higher transcriptional activity | |
| TCF4 FL/Δ | TCF4 FL/Δ: no correlation with transcriptional activity | |
Intron retention | TMyoD1α/β | NA |
| ID1A/B | ID1A: inhibits the formation of E12/MYOD heterodimers.
ID1B: prevents the assembly of E12/E12 homodimers. | |
| ID3/3L | ID3L: reduced antagonistic activity versus E47 | |
Mutually exclusive exons | MEF2A/B/C α1/α2 | MEF2A/B/C α1: repressor of terminal differentiation MEF2A/B/C α2: activator of terminal differentiation |
| TCF4 A-R | TCF4 AD1-containing isoforms: higher transcriptional activity | |
Alternative 5’ or 3’ splice site usage | TCF4 A-R −/+ | TCF4 -/+: no correlation with transactivation activity |
| MEF2C γ | MEF2C γ: reduced transcriptional activity | |
Alternative 3’ terminal exon | CiMDFa/b | CiMDFb: higher myogenic effector function |
| PAX7A/B | PAX7A/B: myogenic commitment | |
| PAX3A/B | NA | |
| PAX3C/D | PAX3C: necessary for terminal myogenic differentiation
PAX3D: maintenance and/or proliferation of muscle precursor | |
Premature termination codon (PTC) | TMyoD1γ | TMyoD1γ: role in the regulation of TmyoD1 expression |

{kind=link}
| Family genes | Transcription factor | Splicing isoforms | Spliced region/domain | Species | References |
|---|---|---|---|---|---|
| Pax | PAX3 | Pax3Q+/Q− | Alternative splicing intron2/exon3 (N-terminus, linker region of the PD) | Zebrafish Mouse Human | [136,137,138,139,140,141] |
| Pax3A/B | Termination at intron 4 (lack of HD and TA domains) | Mouse Human | [148] | ||
| Pax3C/Pax3D | Alternative splicing of exon 8 (C-terminus) | Mouse Human | [146,149] | ||
| PAX7 | Pax7Q+/Q− | Alternative splicing intron2/exon3 (N-terminus, linker region of the PD) | Mouse Human | [146,147] | |
| Pax7GL+/GL− | Alternative splice at intron 3/exon 4 boundary (C-terminus PD subdomain) | Mouse Human | [137,139] | ||
| Pax7A Pax7B | Termination at intron 8 Termination at exon9 (C-terminus) | Mouse Human | [146,147] | ||
| Pax7-2 | Alternative splicing of exon8 (C-terminus) | Chicken | [150] | ||
| bHLH (Class I and II) | MYOD | TMyoD1α/β/γ | Alternative retention of intron I and partial intron II | Takifugu rupripes | [151] |
| CiMDFa/b | Extension to 3’UTR (Domain III) | Ciona intestinalis | [152] | ||
| E2A/TCF3 | E12/E47 E2A-2/5 | Alternative inclusion of exon 15 (bHLH encoding exons - C-terminus) Exclusion of exons 3 and 4 (part of N-terminal AD1) | Mouse Human | [191,192,193] | |
| HEB | Hebα/β | Inclusion of alternate exon (24-amino-acid ankyrin-like motif) | Rat Mouse Human | [194,195] | |
| E2-2/TCF4 | Tcf4A-R Tcf4+/− FL/ΔTcf4 | Mutually exclusive 5’ exons (N-terminus) Alternative splice donor site selection at exon 18 (C-terminus) Exclusion/inclusion exons 8/9 (N-terminal NLS) | Mouse Human | [ 196,197] | |
| HLH (Class V) | ID1 | Id1A/B | Inclusion/exclusion intron I (C-terminus) | Rat Mouse Human | [199] |
| ID3 | Id3/3L | Inclusion/exclusion intron I (C-terminus) | Rat Mouse Human | [200] | |
| Nfi | NFIX | m/h/ha/ch/r Nfix1 m Nfix2 m Nfix3 | Exon9 splicing Exon 7 and 9 splicing Exon 7 and 9 retention (C-terminus) | Chicken Hamster Mouse Rat Human | [73,159] |
| Mef | MEF2A/C/D | Mef2aα1/α2 Mef2cα1/α2 Mef2dα1/α2 Mef2aβ Mef2cβ Mef2dβ Mef2cβ/γ | Alternative splicing exon α1/α2 Exclusion/inclusion exon β Exclusion/inclusion exon β or γ region | Mouse Human | [163,164,165,166,74,173,174,182] [167,175] [168,176,177,178,179] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imbriano, C.; Molinari, S. Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology. Genes 2018, 9, 107. https://doi.org/10.3390/genes9020107
Imbriano C, Molinari S. Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology. Genes. 2018; 9(2):107. https://doi.org/10.3390/genes9020107
Chicago/Turabian StyleImbriano, Carol, and Susanna Molinari. 2018. "Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology" Genes 9, no. 2: 107. https://doi.org/10.3390/genes9020107
APA StyleImbriano, C., & Molinari, S. (2018). Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology. Genes, 9(2), 107. https://doi.org/10.3390/genes9020107