Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors

Abstract
1. Introduction
2. Histone Modifications
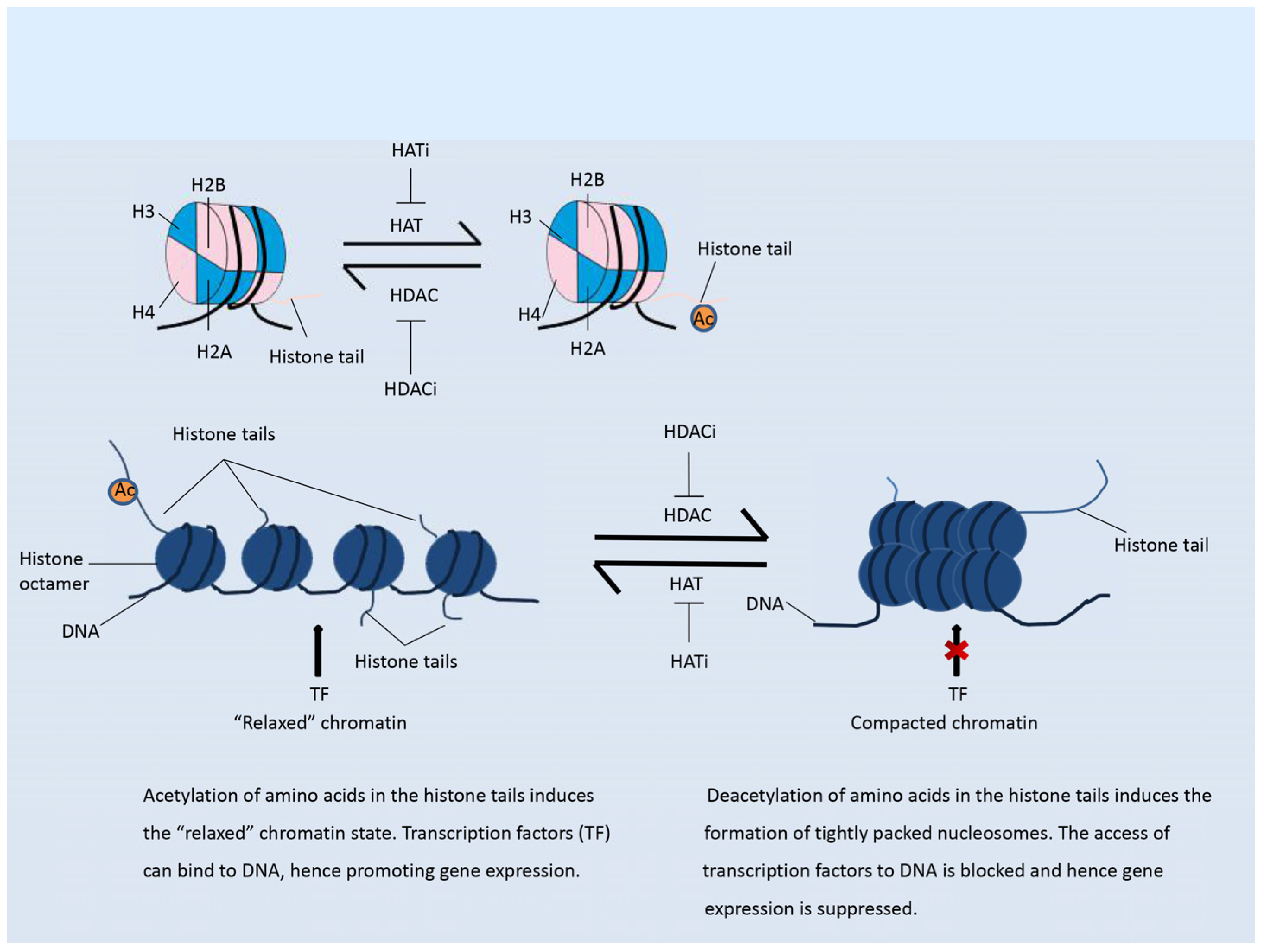
2.1. Histone Acetylation Modifiers
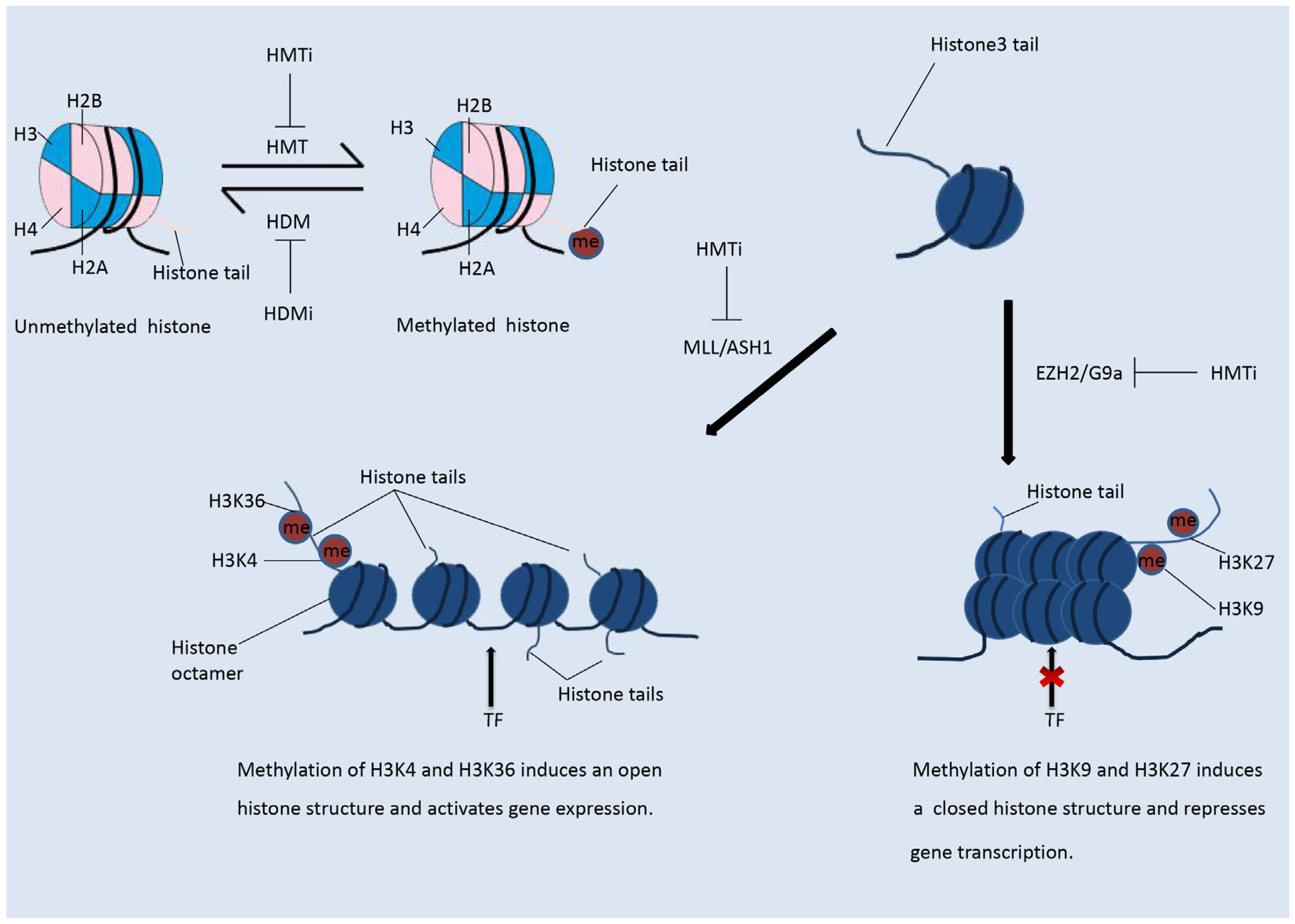
2.2. Histone Methylation Modifiers
2.3. Histone Phosphorylation and Ubiquitination Modifiers
3. Modulation of the Immune Response by Histone Modification
3.1. The Role of the Immune System in Tumorigenesis and Tumor Cell Escape
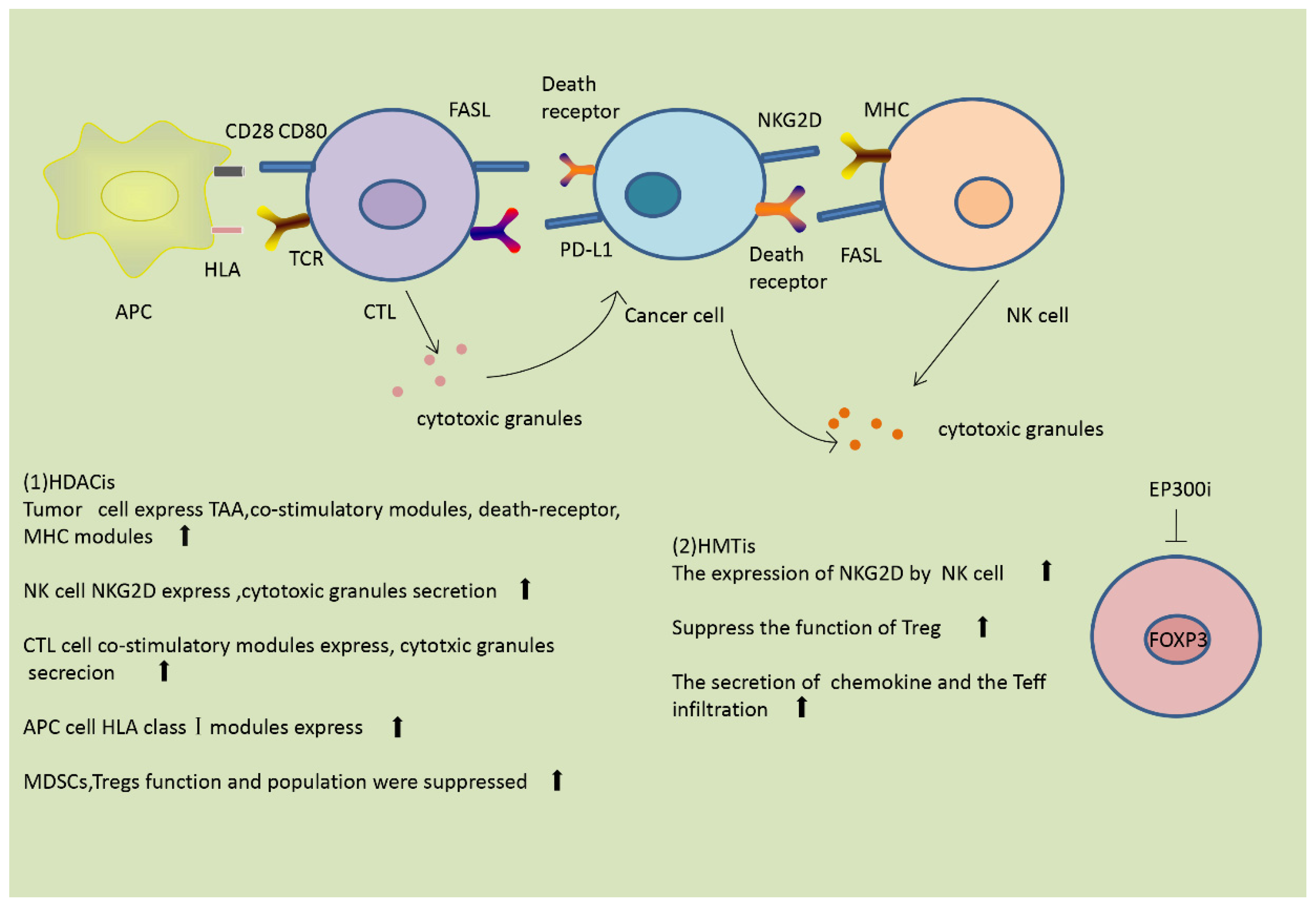
3.2. Effect of Histone Modification on the Immune Response
3.2.1. Modulation of the Immune Response by Histone Acetylation
3.2.2. Modulation of the Immune Response by Histone Methylation
4. Histone Modification Inhibitors Enhance Immune Therapy Efficacy
4.1. Immune Therapy and Cancer
4.2. HDAC Inhibitors in Combination with Immunotherapies in Pre-Clinical and Clinical Trials
4.2.1. HDAC Inhibitors Combined with Immune Checkpoint Inhibitors in Pre-Clinical and Clinical Trials
4.2.2. HDAC Inhibitors Combined with Other Immunotherapies
4.3. Investigation of Histone Methylation Modification Inhibitors in Combination with Immunotherapies in Pre-Clinical and Clinical Trials
4.3.1. Histone Methylation Modification Inhibitors in Combination with Immune Checkpoint Inhibitors in Pre-Clinical and Clinical Trials
4.3.2. Histone Methylation Modulation Inhibitors in Combination with Other Immunotherapies in Pre-Clinical and Clinical Trials
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Spannhoff, A.; Sippl, W.; Jung, M. Cancer treatment of the future: Inhibitors of histone methyltransferases. Int. J. Biochem. Cell Biol. 2009, 41, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Zhou, W.; Shindiapina, P.; Sif, S.; Li, C.; Baiocchi, R.A. Recent advances in targeting protein arginine methyltransferase enzymes in cancer therapy. Expert Opin. Ther. Targets 2018, 22, 527–545. [Google Scholar] [CrossRef] [PubMed]
- De, A.G.; Pienta, K.J. Epigenetic control of macrophage polarization: Implications for targeting tumor-associated macrophages. Oncotarget 2018, 9, 20908. [Google Scholar]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: Targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; De, R.P.; Altucci, L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. Int. J. Biochem. Cell Biol. 2018, 98, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, J.; Chen, Z.; Cheng, A.S. Understanding the epigenetic regulation of tumours and their microenvironments: Opportunities and problems for epigenetic therapy. J. Pathol. 2017, 241, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenet. 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M. Epigenetic Strategies to Boost Cancer Immunotherapies. Int. J. Mol. Sci. 2017, 18, 1108. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.C.; Allis, C.D. Histone methylation versus histone acetylation: New insights into epigenetic regulation. Curre. Opin. Cell Biol. 2001, 13, 263–273. [Google Scholar] [CrossRef]
- Kelly, A.D.; Issa, J.J. The promise of epigenetic therapy: Reprogramming the cancer epigenome. Curr. Opin. Genet. Dev. 2017, 42, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jiao, J.; Wang, L.; O’brien, S.; Newick, K.; Wang, L.C.; Falkensammer, E.; Liu, Y.; Han, R.; Kapoor, V.; et al. HDAC5 controls the functions of Foxp3+ T-regulatory and CD8+ T cells. Int. J. Cancer 2015, 138, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta 2018, 1861, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Beier, U.H.; Akimova, T.; Dahiya, S.; Han, R.; Samanta, A.; Levine, M.H.; Hancock, W.W. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function post-transplantation. Am. J. Transplant. 2018, 18, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Valerio, D.G.; Armstrong, S.A. Exploiting the Epigenome to Control Cancer-Promoting Gene-Expression Programs. Cancer Cell 2016, 29, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, M.G.; Tsatsanis, C.; Kampranis, S.C. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J. Cell. Physiol. 2018, 233, 6495–6507. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, S.N.; Tollefsbol, T.O. Epigenetic Approaches to Cancer Therapy. Epigenet. Hum. Dis. 2018, 6, 219–247. [Google Scholar]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.G.; Bayarsaihan, D. A Novel Epi-drug Therapy Based on the Suppression of BET Family Epigenetic Readers. Yale J. Biol. Med. 2017, 90, 63–71. [Google Scholar] [PubMed]
- Saloura, V.; Vougiouklakis, T.; Sievers, C.; Burkitt, K.; Nakamura, Y.; Hager, G.; van Waes, C. The role of protein methyltransferases as potential novel therapeutic targets in squamous cell carcinoma of the head and neck. Oral Oncol. 2018, 81, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Helin, K.; Minucci, S. The Role of Chromatin-Associated Proteins in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 355–377. [Google Scholar] [CrossRef]
- Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Epigenetic control of the tumor microenvironment. Epigenomics 2016, 12, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Protein methyltransferase inhibitors as precision cancer therapeutics: A decade of discovery. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170080. [Google Scholar] [CrossRef] [PubMed]
- Vanzan, L.; Sklias, A.; Herceg, Z.; Murr, R. Mechanisms of Histone Modifications. In Handbook of Epigenetics; Academic Press: Cambridge, MA, USA, 2017; Chapter 3; pp. 25–45. [Google Scholar]
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics 2016, 8, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Dammeijer, F.; Lau, S.P.; van Eijck, C.H.; van der Burg, S.H.; Aerts, J.G. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017, 36, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Rao, S. Epigenetics and immunotherapy: The current state of play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Covre, A.; Fratta, E.; Di Giacomo, A.M.; Taverna, P.; Natali, P.G.; Coral, S.; Sigalotti, L. Molecular Pathways: At the Crossroads of Cancer Epigenetics and Immunotherapy. Clin. Cancer Res. 2016, 21, 4040–4047. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Thomas, S.; Munster, P.N. Epigenetic modulation with histone deacetylase inhibitors in combination with immunotherapy. Epigenomics 2015, 7, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Tamada, K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015, 106, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Niyongere, S.; Saltos, A.; Gray, J.E. Immunotherapy combination strategies (non-chemotherapy) in non-small cell lung cancer. J. Thorac. Dis. 2018, 10, S433–S450. [Google Scholar] [CrossRef] [PubMed]
- Rodríguezubreva, J.; Garciagomez, A.; Ballestar, E. Epigenetic mechanisms of myeloid differentiation in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Shklovskaya, E.; Hersey, P. Epigenetic modulation in cancer immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Shah, K.; Patel, S.; Jain, N.; Rawal, R. Natural Compounds as Epigenetic Regulators of Human Dendritic Cell-mediated Immune Function. J. Immunother. 2018, 41, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kejík, Z.; Jakubek, M.; Kaplánek, R.; Králová, J.; Mikula, I.; Martásek, P.; Král, V. Epigenetic agents in combined anticancer therapy. Future Med. Chem. 2018, 10, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Subramanian, S. Intrinsic Resistance of Solid Tumors to Immune Checkpoint Blockade Therapy. Cancer Res. 2017, 77, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Jin, T.; Zhu, Y.; Dai, C. Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. 2018, 37, 110. [Google Scholar] [CrossRef] [PubMed]
- Arenas-Ramirez, N.; Sahin, D.; Boyman, O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell. Mol. Life Sci. 2018, 75, 4163–4176. [Google Scholar] [CrossRef] [PubMed]
- Maleki, V.S.; Garrigós, C.; Duran, I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Yao, H.; Li, C.; Fang, J.Y.; Xu, J. Regulation of PD-L1: Emerging Routes for Targeting Tumor Immune Evasion. Curr. Drug Targets 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.; Wherry, E.J. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 2016, 165, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Oezkan, F.; Koenig, M.; Otterson, G.A.; Herman, J.G.; He, K. Epigenetic Therapeutics and Their Impact in Immunotherapy of Lung Cancer. Curr. Pharmacol. Rep. 2017, 3, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Dong, W.; Zhang, C.; Saren, G.; Geng, P.; Zhao, H.; Li, Q.; Zhu, J.; Li, G.; Zhang, S.; et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur. J. Med. Res. 2013, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of Th1 type chemokines shapes tumor immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Chen, J.; Zhao, H.; Zhang, X.; Sharma, P. Epigenetic changes in T cells in response to immune checkpoint blockade. Cancer Res. 2016, 76, 4026. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 |
| Identifier | Recruitment Status | Phase | Cancer Type | Immune Checkpoint Inhibitors | Epigenetic Drugs |
|---|---|---|---|---|---|
| NCT02437136 | Recruiting | I/II | NSCLC and melanoma | Pembrolizumab | Entinostat |
| NCT03179930 | Recruiting | II | Lymphoma Relapsed Refractory | Pembrolizumab | Entinostat |
| NCT02909452 | Active, not recruiting | I | Advanced solid tumors | Pembrolizumab | Entinostat |
| NCT02395627 | Recruiting | II | Breast Neoplasms | Pembrolizumab | Vorinostat |
| NCT02619253 | Recruiting | I | Renal Cell Carcinoma, Urinary Bladder Neoplasms | Pembrolizumab | Vorinostat |
| NCT02538510 | Active, not recruiting | I/II | Recurrent unresectable/metastatic HNSCC and SGC | Pembrolizumab | Vorinostat |
| NCT02512172 | Recruiting | I | Colorectal Cancer | Pembrolizumab | Romidepsin with or without azacytidine(DNMTi) |
| NCT02697630 | Recruiting | II | Metastatic Uveal Melanoma | Pembrolizumab | Entinostat |
| NCT02453620 | Recruiting | I | Breast Adenocarcinoma HER2/Neu Negative Invasive Breast Carcinoma | Ipilimumab | Entinostat |
| NCT03552380 | Recruiting | II | Renal Cell Carcinoma | Pembrolizumab plus Ipilimumab | Entinostat |
| NCT03250273 | Recruiting | II | Cholangiocarcinoma and Pancreatic Cancer and Metastatic Pancreatic Cancer | Nivolumab | Entinostat |
| NCT03278782 | Recruiting | I/II | Lymphoid Haematopoietic Malignant Neoplasms Cutaneous T-Cell Lymphoma Refractory Cutaneous T-cell Lymphoma | Pembrolizumab | Romidepsin |
| NCT03150329 | Recruiting | I | Relapsed or Refractory Diffuse Large B-Cell Lymphoma, Follicular Lymphoma, or Hodgkin Lymphoma | Pembrolizumab | Vorinostat |
| NCT02915523 | Active, not recruiting | I/II | Epithelial Ovarian Cancer, Peritoneal Cancer, Fallopian Tube Cancer | Avelumab | Entinostat |
| NCT02708680 | Recruiting | I/II | Breast Cancer | Atezolizumab | Entinostat |
| NCT02638090 | Recruiting | I/II | Lung Cancer, Non-small Cell Lung Cancer | Pembrolizumab | Vorinostat |
| NCT02220842 | Active, not recruiting | I | Lymphoma | Atezolizumab administered with Obinutuzumab | Tazemetostat |
| NCT03525795 | Recruiting | I/II | Advanced Solid Tumors | Ipilimumab | CPI-1205 |
| NCT02032810 | Active, not recruiting | I | Melanoma/Skin Cancer | Ipilimumab | Panobinostat |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, W.; Lv, S.; Li, H.; Cui, W.; Wang, L. Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors. Genes 2018, 9, 633. https://doi.org/10.3390/genes9120633
Sun W, Lv S, Li H, Cui W, Wang L. Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors. Genes. 2018; 9(12):633. https://doi.org/10.3390/genes9120633
Chicago/Turabian StyleSun, Wanyu, Shuting Lv, Hong Li, Wei Cui, and Lihui Wang. 2018. "Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors" Genes 9, no. 12: 633. https://doi.org/10.3390/genes9120633
APA StyleSun, W., Lv, S., Li, H., Cui, W., & Wang, L. (2018). Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors. Genes, 9(12), 633. https://doi.org/10.3390/genes9120633