RAD-ical New Insights into RAD51 Regulation


Abstract
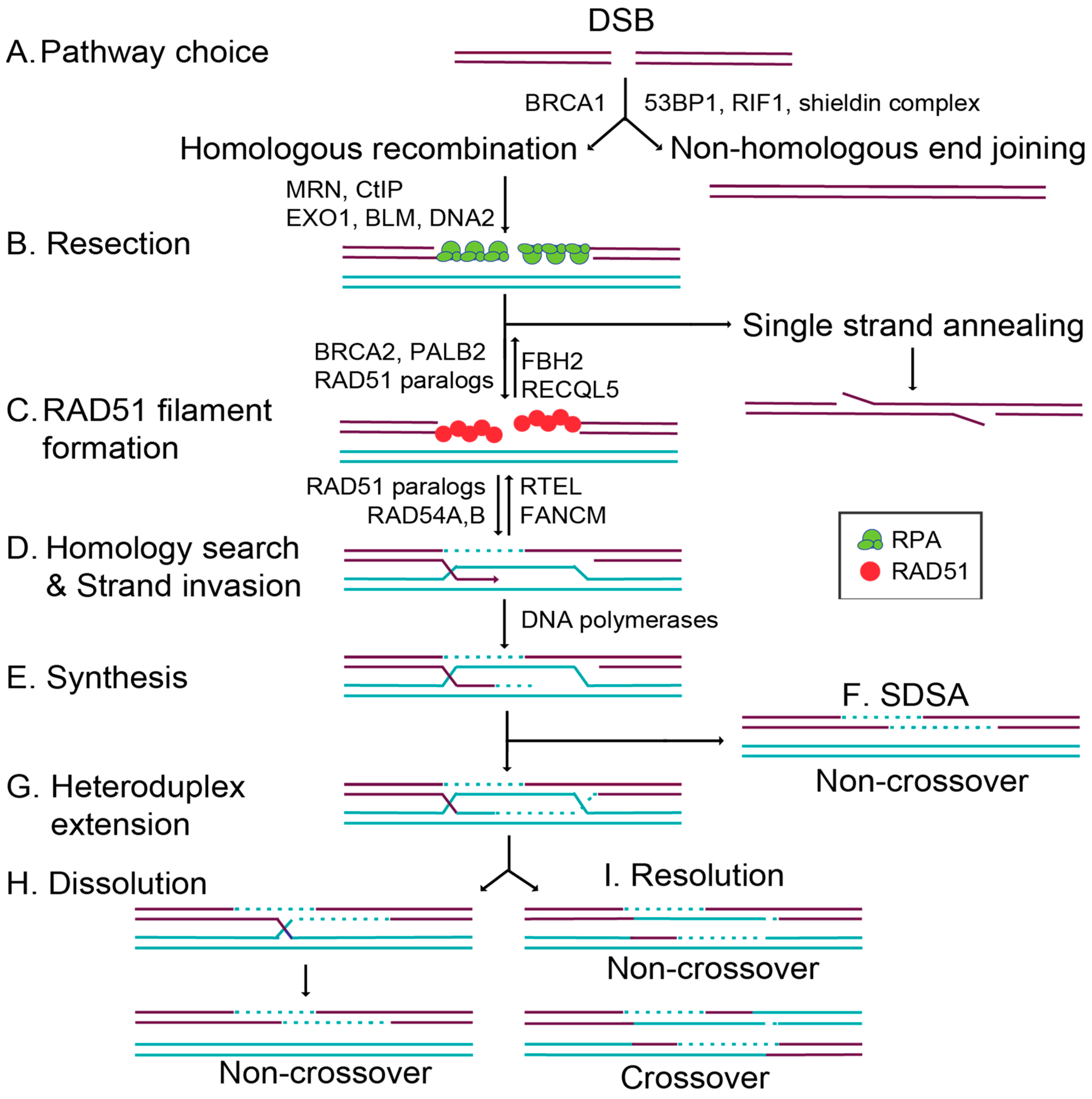
1. Introduction to Double-Strand Break and Repair
1.1. Commitment to Homolgous Recombination through Double-Strand Break End Resection and RAD51 Filament Formation
1.2. Overview of RAD51 Structure, Function, and Activity
1.3. Overview of Key RAD51 Regulators
2. RAD51 Regulation in Mammalian Models
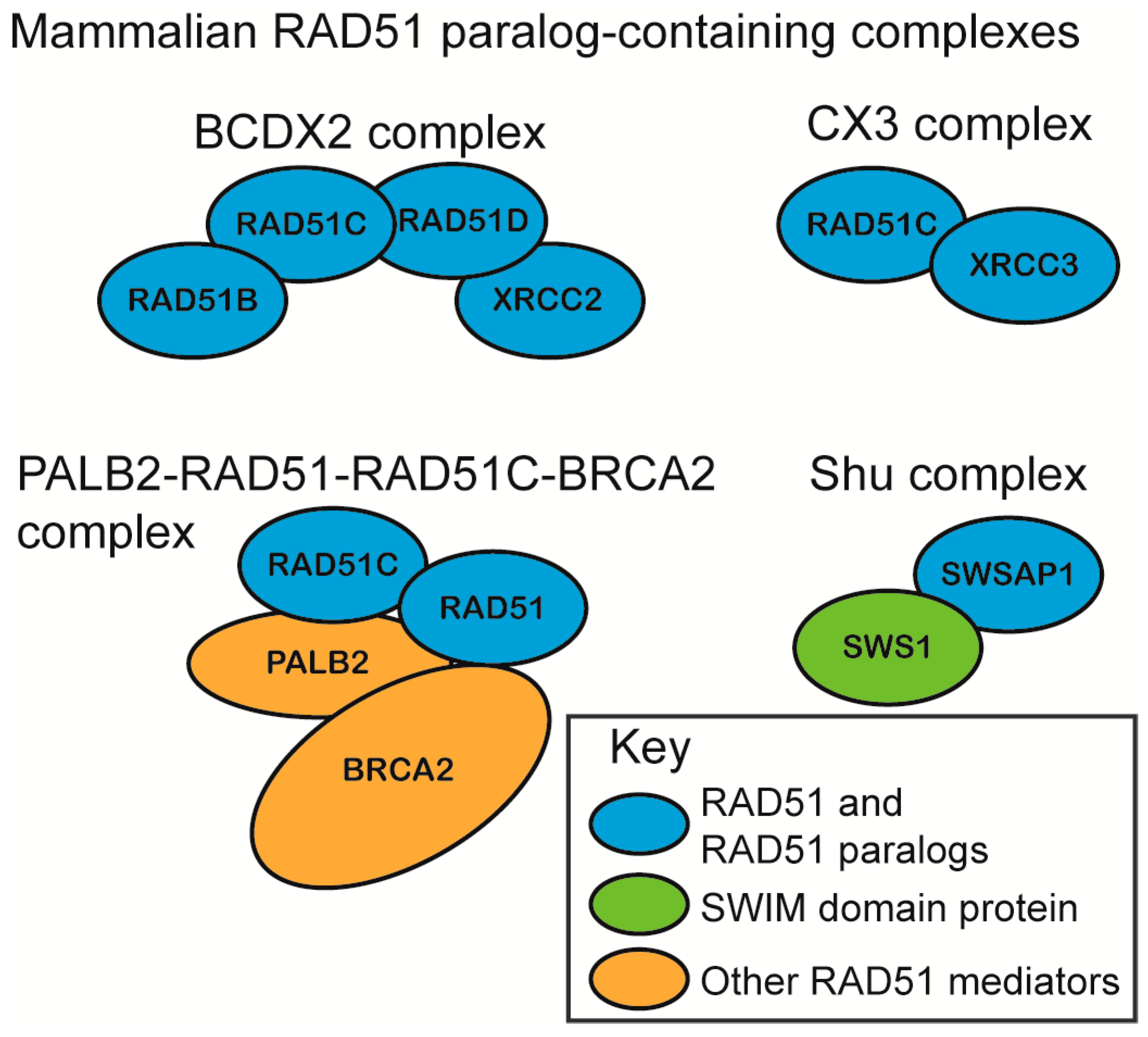
2.1. RAD51 Paralog Containing Complexes
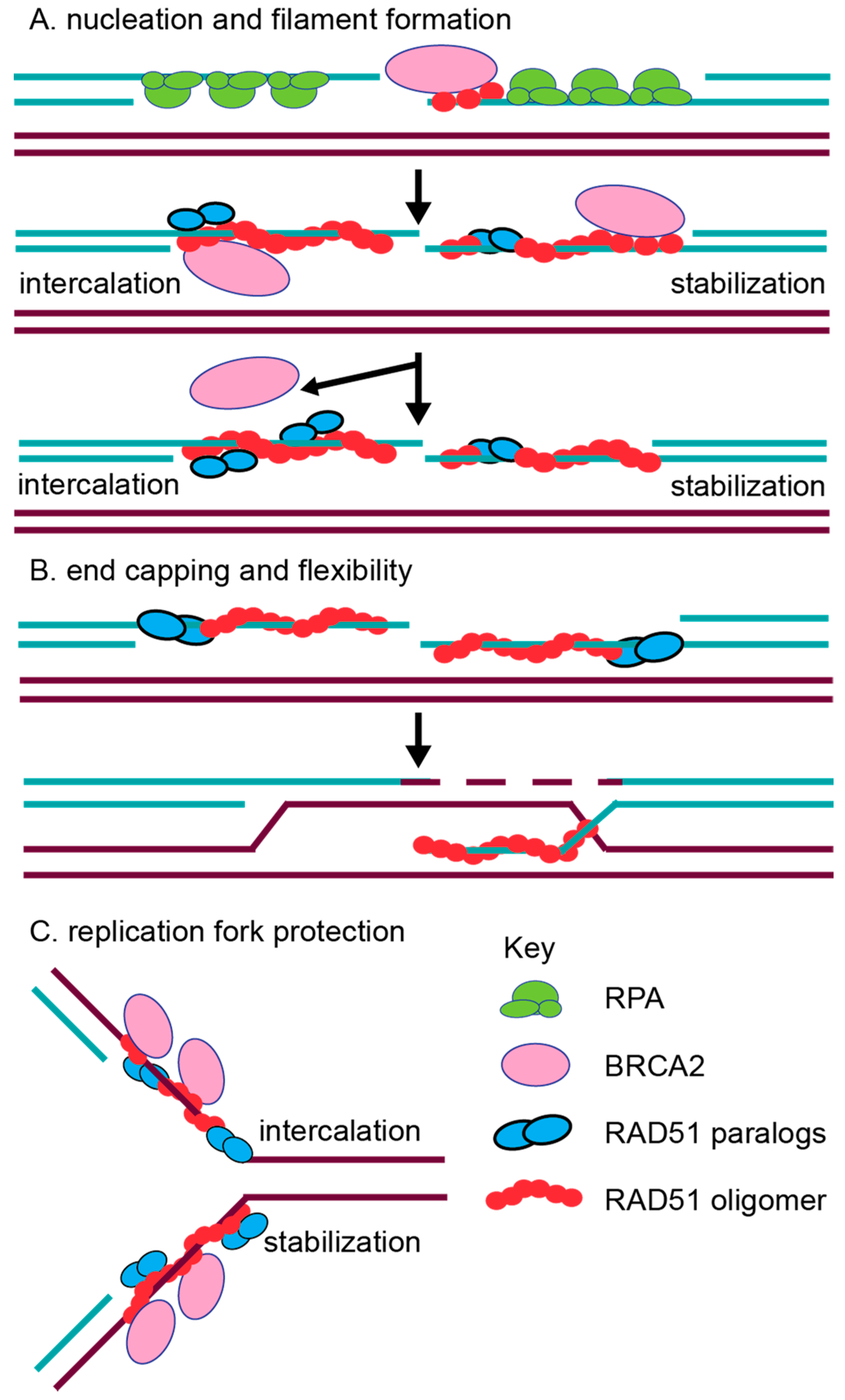
2.2. In Vitro Characterization of RAD51 Paralog Function in RAD51 Pre-Synaptic and Post-Synaptic Filament Assembly
2.3. In Vivo Characterization of RAD51 Paralog Function in Vertebrates
2.3.1. RAD51 Paralog Knockout Mice and Mouse Embryonic Fibroblasts
2.3.2. RAD51 Paralog Knockout Hamster, Chicken, and Tumor Cell Lines
2.4. The RAD51 Paralogs Function at Replication Forks
3. RAD51 Mediators and Disease
3.1. Genetic Syndromes Linked to RAD51 Mediators
3.2. Cancers Associated with Defects in Homologous Recombination
3.3. Therapeutic Strategies for Homologous Recombination-Deficient Breast and Ovarian Cancers
4. Synthetic Lethality in BRCA and BRCA-Like Cancers
5. Chemotherapeutics Currently in the Clinic
6. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. 2014, 26, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Greenberg, R.A. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016, 30, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.A.; Rothstein, R. At loose ends: Resecting a double-strand break. Cell 2009, 137, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Spier, I.; Kerick, M.; Drichel, D.; Horpaopan, S.; Altmuller, J.; Laner, A.; Holzapfel, S.; Peters, S.; Adam, R.; Zhao, B.; et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam. Cancer 2016, 15, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.K.; Sullivan, M.R.; Bernstein, K.A. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem. Cell Biol. 2016, 94, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Gutierrez-Enriquez, S.; Santamarina, M.; Montalban, G.; Bonache, S.; Balmana, J.; Carracedo, A.; Diez, O.; Vega, A. RAD51C germline mutations found in Spanish site-specific breast cancer and breast-ovarian cancer families. Breast Cancer Res. Treat. 2014, 147, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Kong, H.; Nei, M.; Ma, H. Origins and evolution of the recA/RAD51 gene family: Evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 10328–10333. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA sequence alignment by microhomology sampling during homologous recombination. Cell 2015, 160, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Terakawa, T.; Qi, Z.; Steinfeld, J.B.; Redding, S.; Kwon, Y.; Gaines, W.A.; Zhao, W.; Sung, P.; Greene, E.C. DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science 2015, 349, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Rothstein, R. Poetry in motion: Increased chromosomal mobility after DNA damage. DNA Repair 2017, 56, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Mine-Hattab, J.; Rothstein, R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 2012, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007, 9, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Soutoglou, E.; Misteli, T. Mobility and immobility of chromatin in transcription and genome stability. Curr. Opin. Genet. Dev. 2007, 17, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.M.; Borovski, T.; Stap, J.; Cijsouw, T.; ten Cate, R.; Medema, J.P.; Kanaar, R.; Franken, N.A.; Aten, J.A. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 2012, 125, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.S.; Hasty, P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 1996, 16, 7133–7143. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Fujii, Y.; Sakumi, K.; Tominaga, Y.; Nakao, K.; Sekiguchi, M.; Matsushiro, A.; Yoshimura, Y.; Morita, T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6236–6240. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.; Spirek, M.; Chaurasiya, K.R.; Ward, J.D.; Carzaniga, R.; Yu, X.; Egelman, E.H.; Collinson, L.M.; Rueda, D.; Krejci, L.; et al. Rad51 Paralogs Remodel Pre-synaptic Rad51 Filaments to Stimulate Homologous Recombination. Cell 2015, 162, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.G.; Spirek, M.; Jian Ma, C.; Carzaniga, R.; Takaki, T.; Collinson, L.M.; Greene, E.C.; Krejci, L.; Boulton, S.J. A Polar and Nucleotide-Dependent Mechanism of Action for RAD51 Paralogs in RAD51 Filament Remodeling. Mol. Cell 2016, 64, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, H.; Paul, M.W.; Grosbart, M.; van Rossum-Fikkert, S.E.; Lebbink, J.H.G.; Kanaar, R.; Houtsmuller, A.B.; Wyman, C. Architectural plasticity of human BRCA2-RAD51 complexes in DNA break repair. Nucleic Acids Res. 2017, 45, 4507–4518. [Google Scholar] [CrossRef] [PubMed]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Mangion, J.; Russell, P.; Seal, S.; Barfoot, R.; Ponder, B.A.; Stratton, M.R.; Easton, D. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat. Genet. 1997, 15, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef] [PubMed]
- Wesoly, J.; Agarwal, S.; Sigurdsson, S.; Bussen, W.; Van Komen, S.; Qin, J.; van Steeg, H.; van Benthem, J.; Wassenaar, E.; Baarends, W.M.; et al. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol. Cell. Biol. 2006, 26, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, A.; Mazin, A.; Kowalczykowski, S.C. Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat. Struct. Mol. Biol. 2003, 10, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Rothstein, R. Cell biology of mitotic recombination. Cold Spring Harbor Perspect. Biol. 2015, 7, a016535. [Google Scholar] [CrossRef] [PubMed]
- Karpenshif, Y.; Bernstein, K.A. From yeast to mammals: Recent advances in genetic control of homologous recombination. DNA Repair 2012, 11, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Simandlova, J.; Zagelbaum, J.; Payne, M.J.; Chu, W.K.; Shevelev, I.; Hanada, K.; Chatterjee, S.; Reid, D.A.; Liu, Y.; Janscak, P.; et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J. Biol. Chem. 2013, 288, 34168–34180. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.J.; Cox, R.; Thacker, J. Isolation and cross-sensitivity of X-ray-sensitive mutants of V79-4 hamster cells. Mutat. Res. 1987, 183, 279–286. [Google Scholar] [CrossRef]
- Thacker, J. A surfeit of RAD51-like genes? Trends Genet. 1999, 15, 166–168. [Google Scholar] [CrossRef]
- Cartwright, R.; Tambini, C.E.; Simpson, P.J.; Thacker, J. The XRCC2 DNA repair gene from human and mouse encodes a novel member of the recA/RAD51 family. Nucleic Acids Res. 1998, 26, 3084–3089. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Schild, D. The contribution of homologous recombination in preserving genome integrity in mammalian cells. Biochimie 1999, 81, 87–105. [Google Scholar] [CrossRef]
- Adam, J.; Deans, B.; Thacker, J. A role for Xrcc2 in the early stages of mouse development. DNA Repair 2007, 6, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wan, L.; Wu, Y.; Chen, J.; Huang, J. hSWS1.SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair. J. Biol. Chem. 2011, 286, 41758–41766. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.Y.; Stasiak, A.Z.; Stasiak, A.; Benson, F.E.; West, S.C. Complex formation by the human RAD51C and XRCC3 recombination repair proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 8440–8446. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.Y.; Tarsounas, M.C.; Stasiak, A.Z.; Stasiak, A.; Shah, R.; McIlwraith, M.J.; Benson, F.E.; West, S.C. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev. 2001, 15, 3296–3307. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Chahwan, C.; Gao, H.; Blais, V.; Wohlschlegel, J.; Yates, J.R., 3rd; McGowan, C.H.; Russell, P. Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J. 2006, 25, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- McClendon, T.B.; Sullivan, M.R.; Bernstein, K.A.; Yanowitz, J.L. Promotion of Homologous Recombination by SWS-1 in Complex with RAD-51 Paralogs in Caenorhabditis elegans. Genetics 2016, 203, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Martino, J.; Bernstein, K.A. The Shu complex is a conserved regulator of homologous recombination. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Schild, D.; Lio, Y.C.; Collins, D.W.; Tsomondo, T.; Chen, D.J. Evidence for simultaneous protein interactions between human Rad51 paralogs. J. Biol. Chem. 2000, 275, 16443–16449. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Yoshikawa, D.M.; McConnell, I.R.; Clark, R.; Schild, D.; Albala, J.S. RAD51C interacts with RAD51B and is central to a larger protein complex in vivo exclusive of RAD51. J. Biol. Chem. 2002, 277, 8406–8411. [Google Scholar] [CrossRef] [PubMed]
- Wiese, C.; Collins, D.W.; Albala, J.S.; Thompson, L.H.; Kronenberg, A.; Schild, D. Interactions involving the Rad51 paralogs Rad51C and XRCC3 in human cells. Nucleic Acids Res. 2002, 30, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Kurumizaka, H.; Ikawa, S.; Nakada, M.; Eda, K.; Kagawa, W.; Takata, M.; Takeda, S.; Yokoyama, S.; Shibata, T. Homologous-pairing activity of the human DNA-repair proteins Xrcc3.Rad51C. Proc. Natl. Acad. Sci. USA 2001, 98, 5538–5543. [Google Scholar] [CrossRef] [PubMed]
- Braybrooke, J.P.; Spink, K.G.; Thacker, J.; Hickson, I.D. The RAD51 family member, RAD51L3, is a DNA-stimulated ATPase that forms a complex with XRCC2. J. Biol. Chem. 2000, 275, 29100–29106. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.; Van Komen, S.; Bussen, W.; Schild, D.; Albala, J.S.; Sung, P. Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev. 2001, 15, 3308–3318. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell. Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Sawicka, D.; Barsky, D.; Albala, J.S. Domain mapping of the Rad51 paralog protein complexes. Nucleic Acids Res. 2004, 32, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, L.; Tao, Y.; Bai, T.; Lu, R.; Zhang, T.; Chen, J.; Ding, J. Structural basis for the functional role of the Shu complex in homologous recombination. Nucleic Acids Res. 2017, 45, 13068–13079. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Kurumizaka, H.; Ikawa, S.; Nakada, M.; Enomoto, R.; Kagawa, W.; Kinebuchi, T.; Yamazoe, M.; Yokoyama, S.; Shibata, T. Homologous pairing and ring and filament structure formation activities of the human Xrcc2*Rad51D complex. J. Biol. Chem. 2002, 277, 14315–14320. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Sasaki, M.S.; Tachiiri, S.; Fukushima, T.; Sonoda, E.; Schild, D.; Thompson, L.H.; Takeda, S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol. 2001, 21, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Gaines, W.A.; Godin, S.K.; Kabbinavar, F.F.; Rao, T.; VanDemark, A.P.; Sung, P.; Bernstein, K.A. Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat. Commun. 2015, 6, 7834. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997, 11, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.K.; Zhang, Z.; Herken, B.W.; Westmoreland, J.W.; Lee, A.G.; Mihalevic, M.J.; Yu, Z.; Sobol, R.W.; Resnick, M.A.; Bernstein, K.A. The Shu complex promotes error-free tolerance of alkylation-induced base excision repair products. Nucleic Acids Res. 2016, 44, 8199–8215. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.; Prakash, R.; Romanienko, P.J.; Roig, I.; Keeney, S.; Jasin, M. Shu complex SWS1-SWSAP1 promotes early steps in mouse meiotic recombination. Nat. Commun. 2018, 9, 3961. [Google Scholar] [CrossRef] [PubMed]
- Somyajit, K.; Saxena, S.; Babu, S.; Mishra, A.; Nagaraju, G. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 2015, 43, 9835–9855. [Google Scholar] [CrossRef] [PubMed]
- Deans, B.; Griffin, C.S.; Maconochie, M.; Thacker, J. Xrcc2 is required for genetic stability, embryonic neurogenesis and viability in mice. EMBO J. 2000, 19, 6675–6685. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.G.; Haines, D.C.; Martin, B.K.; Sharan, S.K. Loss of Rad51c leads to embryonic lethality and modulation of Trp53-dependent tumorigenesis in mice. Cancer Res. 2009, 69, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Pittman, D.L.; Schimenti, J.C. Midgestation lethality in mice deficient for the RecA-related gene, Rad51d/Rad51l3. Genesis 2000, 26, 167–173. [Google Scholar] [CrossRef]
- Shu, Z.; Smith, S.; Wang, L.; Rice, M.C.; Kmiec, E.B. Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can Be partially rescued in a p53−/− background. Mol. Cell. Biol. 1999, 19, 8686–8693. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R.; de la Pompa, J.L.; Mak, T.W. Developmental studies of Brca1 and Brca2 knock-out mice. J. Mammary Gland Biol. Neoplasia 1998, 3, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Deans, B.; Griffin, C.S.; O’Regan, P.; Jasin, M.; Thacker, J. Homologous recombination deficiency leads to profound genetic instability in cells derived from Xrcc2-knockout mice. Cancer Res. 2003, 63, 8181–8187. [Google Scholar] [PubMed]
- Smiraldo, P.G.; Gruver, A.M.; Osborn, J.C.; Pittman, D.L. Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res. 2005, 65, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.; Pellegrini, M.; Shuda, K.; Fernandez-Capetillo, O.; Liu, Y.; Martin, B.K.; Burkett, S.; Southon, E.; Pati, D.; Tessarollo, L.; et al. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J. Cell Biol. 2007, 176, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.; Hanenberg, H.; Schuster, B.; Barker, K.; Wiek, C.; Erven, V.; Neveling, K.; Endt, D.; Kesterton, I.; Autore, F.; et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010, 42, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Jacquinet, A.; Brown, L.; Sawkins, J.; Liu, P.; Pugash, D.; Van Allen, M.I.; Patel, M.S. Expanding the FANCO/RAD51C associated phenotype: Cleft lip and palate and lobar holoprosencephaly, two rare findings in Fanconi anemia. Eur. J. Med. Genet. 2018, 61, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Elfaki, M.; Alkuraya, F.S. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J. Med. Genet. 2012, 49, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [PubMed]
- Johnson, R.D.; Liu, N.; Jasin, M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 1999, 401, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Fukushima, T.; Morrison, C.; Albala, J.S.; Swagemakers, S.M.; Kanaar, R.; Thompson, L.H.; Takeda, S. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell. Biol. 2000, 20, 6476–6482. [Google Scholar] [CrossRef] [PubMed]
- Fuller, L.F.; Painter, R.B. A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutat. Res. 1988, 193, 109–121. [Google Scholar] [CrossRef]
- Hu, T.; Miller, C.M.; Ridder, G.M.; Aardema, M.J. Characterization of p53 in Chinese hamster cell lines CHO-K1, CHO-WBL, and CHL: Implications for genotoxicity testing. Mutat. Res 1999, 426, 51–62. [Google Scholar] [CrossRef]
- Bishop, D.K.; Ear, U.; Bhattacharyya, A.; Calderone, C.; Beckett, M.; Weichselbaum, R.R.; Shinohara, A. Xrcc3 is required for assembly of Rad51 complexes in vivo. J. Biol. Chem. 1998, 273, 21482–21488. [Google Scholar] [CrossRef] [PubMed]
- Lio, Y.C.; Schild, D.; Brenneman, M.A.; Redpath, J.L.; Chen, D.J. Human Rad51C deficiency destabilizes XRCC3, impairs recombination, and radiosensitizes S/G2-phase cells. J. Biol. Chem. 2004, 279, 42313–42320. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Schild, D. Recombinational DNA repair and human disease. Mutat. Res. 2002, 509, 49–78. [Google Scholar] [CrossRef]
- Moldovan, G.L.; D’Andrea, A.D. How the Fanconi Anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi Anemia Signaling and Cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Knies, K.; Inano, S.; Ramirez, M.J.; Ishiai, M.; Surralles, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Ollier, M.; Radosevic-Robin, N.; Kwiatkowski, F.; Ponelle, F.; Viala, S.; Privat, M.; Uhrhammer, N.; Bernard-Gallon, D.; Penault-Llorca, F.; Bignon, Y.J.; et al. DNA repair genes implicated in triple negative familial non-BRCA1/2 breast cancer predisposition. Am. J. Cancer Res. 2015, 5, 2113–2126. [Google Scholar] [PubMed]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Bernards, S.S.; Pennington, K.P.; Harrell, M.I.; Agnew, K.J.; Garcia, R.L.; Norquist, B.M.; Swisher, E.M. Clinical characteristics and outcomes of patients with BRCA1 or RAD51C methylated versus mutated ovarian carcinoma. Gynecol. Oncol. 2018, 148, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, L.M.; Nurminen, R.; Gylfe, A.; Aaltonen, L.A.; Schleutker, J.; Nevanlinna, H. Screening of Finnish RAD51C founder mutations in prostate and colorectal cancer patients. BMC Cancer 2012, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Morganella, S. Mutational Signatures in Breast Cancer: The Problem at the DNA Level. Clin Cancer Res. 2017, 23, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef] [PubMed]
- Vencken, P.M.; Kriege, M.; Hoogwerf, D.; Beugelink, S.; van der Burg, M.E.; Hooning, M.J.; Berns, E.M.; Jager, A.; Collee, M.; Burger, C.W.; et al. Chemosensitivity and outcome of BRCA1- and BRCA2-associated ovarian cancer patients after first-line chemotherapy compared with sporadic ovarian cancer patients. Ann. Oncol. 2011, 22, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Matondo, A.; Jo, Y.H.; Shahid, M.; Choi, T.G.; Nguyen, M.N.; Nguyen, N.N.Y.; Akter, S.; Kang, I.; Ha, J.; Maeng, C.H.; et al. The Prognostic 97 Chemoresponse Gene Signature in Ovarian Cancer. Sci. Rep. 2017, 7, 9689. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Rivera, B.; Di Iorio, M.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally Null RAD51D Missense Mutation Associates Strongly with Ovarian Carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef] [PubMed]
- Kristeleit, R.S.; Miller, R.E.; Kohn, E.C. Gynecologic Cancers: Emerging Novel Strategies for Targeting DNA Repair Deficiency. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e259–e268. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Kaye, S.B.; Yap, T.A. PARP inhibitors: The race is on. Br. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| BCDX2 | RAD51B–RAD51C–RAD51D–XRCC2 complex |
| BC | RAD51B–RAD51C subcomplex |
| BER | Base excision repair |
| BIR | Break-induced replication |
| CHO | Chinese hamster ovary |
| CO | Crossover |
| CX3 | RAD51C–XRCC3 complex |
| DDR | DNA damage response |
| DX2 | RAD51D–XRCC2 subcomplex |
| dHJ | Double Holliday junction |
| DSB | Double-strand break |
| dsDNA | Double-stranded DNA |
| D-loop | Displacement loop |
| FA | Fanconi anemia |
| FANCD1 | BRCA2 |
| FANCN | PALB2 |
| FANCO | RAD51C |
| FANCR | RAD51 |
| FANCU | XRCC2 |
| HR | Homologous recombination |
| ICL | Interstrand crosslink |
| IR | Ionizing radiation |
| LOH | Loss of heterozygosity |
| MEF | Mouse embryonic fibroblast |
| MMC | Mitomycin C |
| MMS | Methylmethane sulfonate |
| MRN | MRE11–RAD51–NBS1 |
| NCO | Non-crossover |
| NHEJ | Non-homologous end joining |
| PARP | Poly (ADP-ribose) polymerase |
| PARPi | Poly (ADP-ribose) polymerase inhibitor |
| SCEs | Sister chromatid exchanges |
| SDSA | Synthesis-dependent strand annealing |
| ssDNA | Single-stranded DNA |
| TIRF | Total internal reflection fluorescence |
| RAD51 Paralog | Complex Member | Mouse Knockout | p53−/− Rescue | MEF MMC Sensitivity | MEF MMS Sensitivity | Other Phenotypes |
|---|---|---|---|---|---|---|
| RAD51B | BCDX2 | E7.5-8.5 [89] | partial [89] | NA | NA | NA |
| RAD51C | BCDX2, CX3, PALB2-RAD51- RAD51C-BRCA2 | E8.5 [95] | partial [87] | 2–3 fold (p53−/−) [87] | 2–3 fold (p53−/−) [87] | ↓SCEs [87] ↓IR-RAD51 foci (p53−/−) [87] |
| RAD51D | BCDX2 | E9-E10 [88] | Partial [92] | 17.6 fold (p53−/−) [92] | 6.3 fold (p53−/−) [92] | ↓SCEs (p53−/−) [92] ↓IR-RAD51 foci (p53−/−) [92] |
| XRCC2 | BCDX2 | E10.5-died at birth [86] | yes died 6d P/N [61] | 4.5 fold (p53+) [91] | NA | ↓SCEs [91] ↓IR-RAD51 foci [91] |
| XRCC3 | CX3 | NA | NA | NA | NA | NA |
| SWSAP1 | Shu Complex (SWS1) | Viable/Infertile [84] | NA | NA | NA | NA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, M.R.; Bernstein, K.A. RAD-ical New Insights into RAD51 Regulation. Genes 2018, 9, 629. https://doi.org/10.3390/genes9120629
Sullivan MR, Bernstein KA. RAD-ical New Insights into RAD51 Regulation. Genes. 2018; 9(12):629. https://doi.org/10.3390/genes9120629
Chicago/Turabian StyleSullivan, Meghan R., and Kara A. Bernstein. 2018. "RAD-ical New Insights into RAD51 Regulation" Genes 9, no. 12: 629. https://doi.org/10.3390/genes9120629
APA StyleSullivan, M. R., & Bernstein, K. A. (2018). RAD-ical New Insights into RAD51 Regulation. Genes, 9(12), 629. https://doi.org/10.3390/genes9120629