The Unresolved Problem of DNA Bridging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
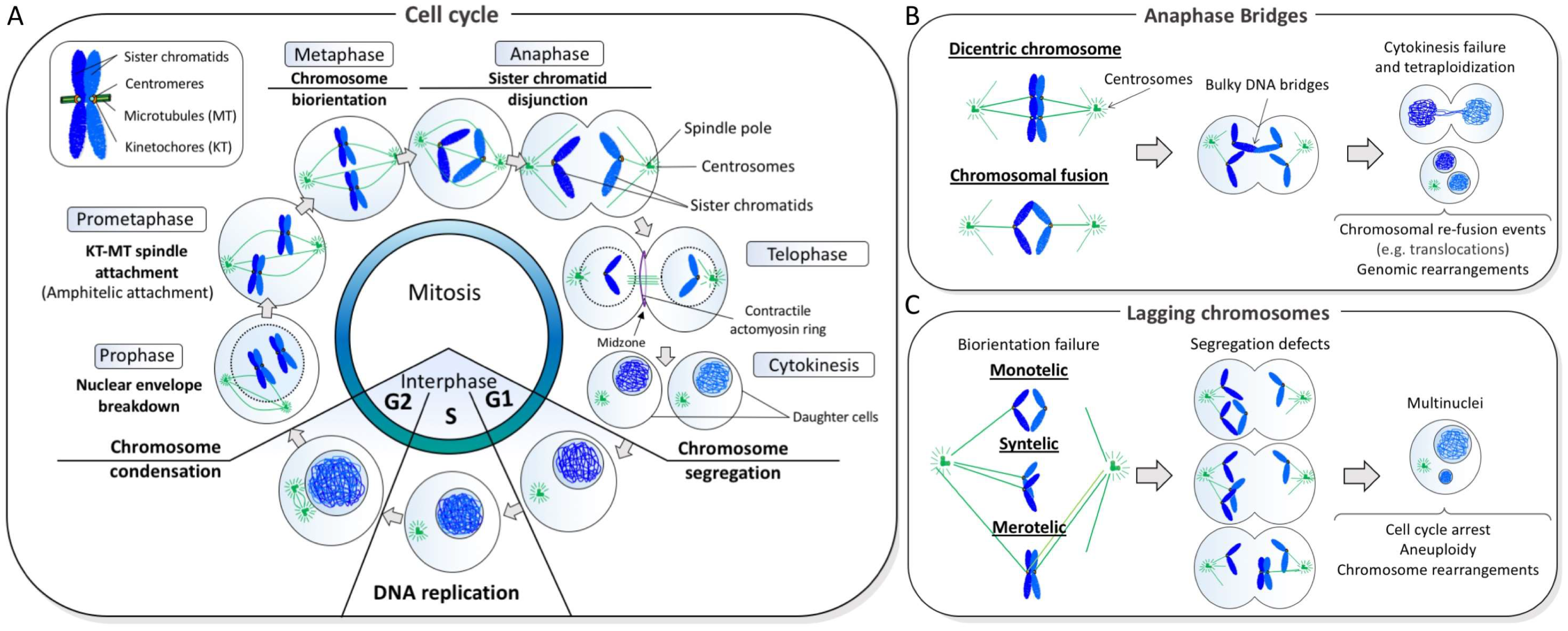
1. Chromosome Mis-Segregation and Genome Instability
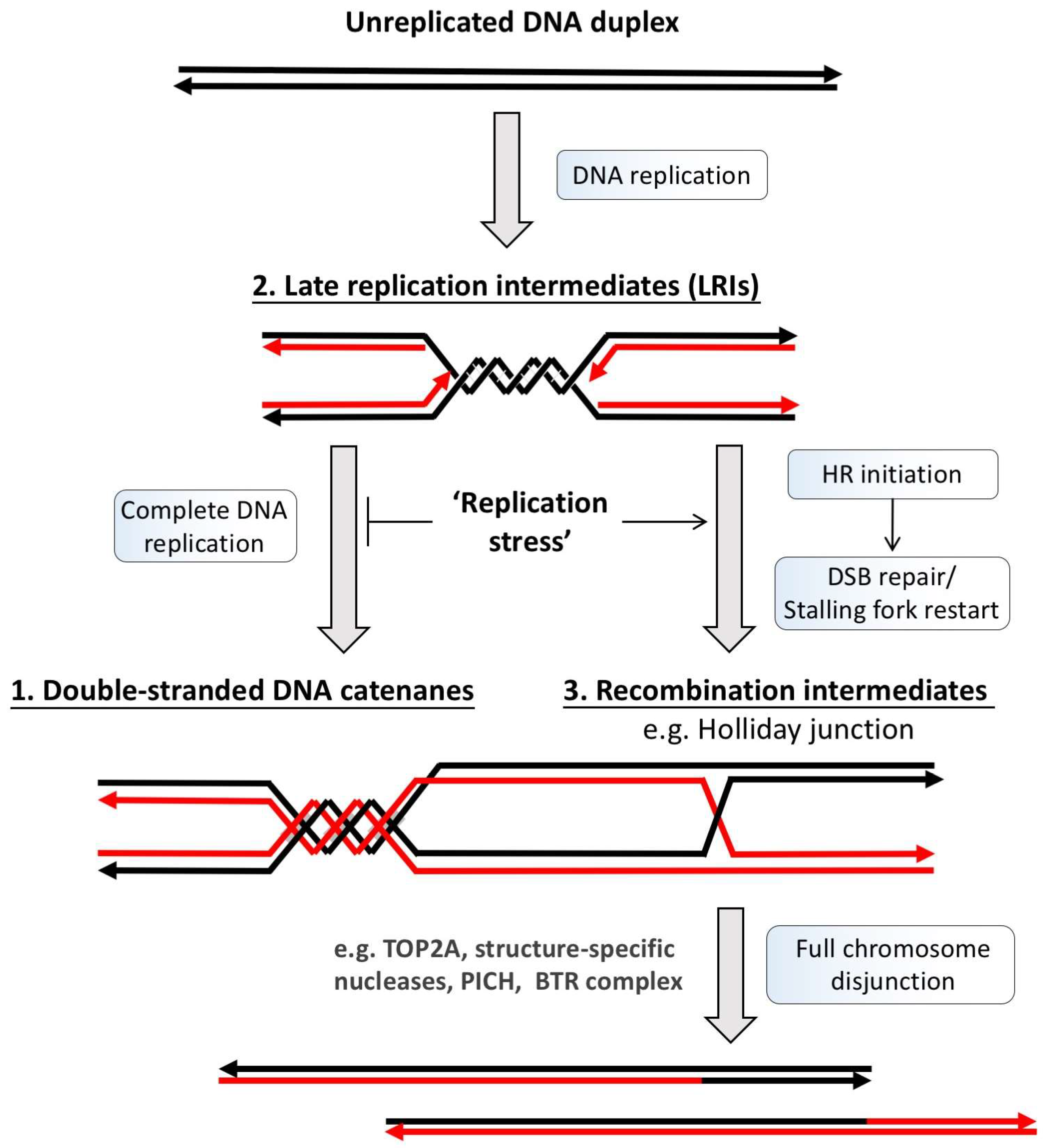
2. Classification of Ultra-Fine DNA Bridges
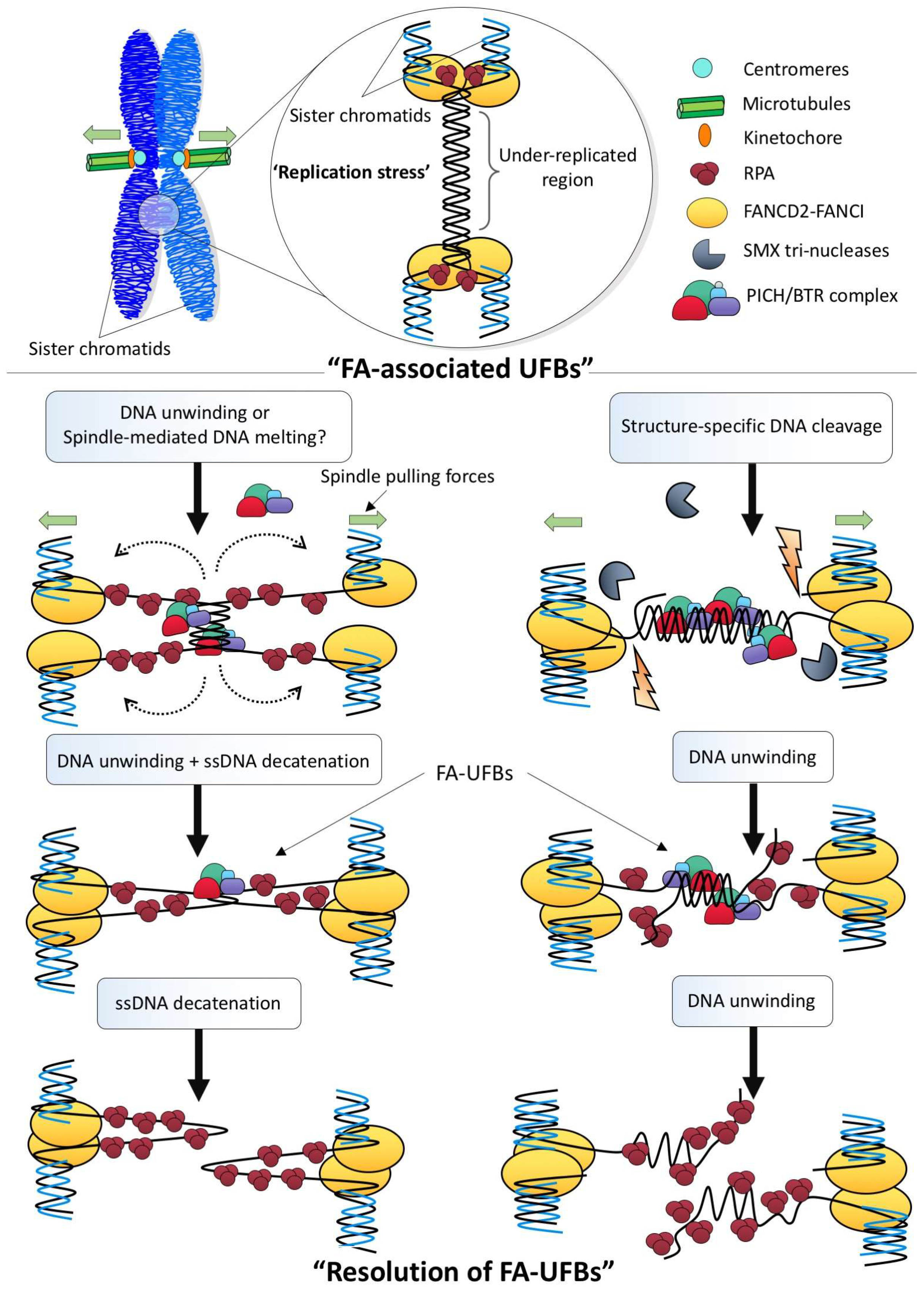
3. Ultra-Fine DNA Bridge-Associated Factors
4. How Are Different Types of Ultra-Fine DNA Bridges Resolved?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Potapova, T.; Gorbsky, G.J. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The Behavior in Successive Nuclear Divisions of a Chromosome Broken at Meiosis. Proc. Natl. Acad. Sci. USA 1939, 25, 405–416. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The Fusion of Broken Ends of Chromosomes Following Nuclear Fusion. Proc. Natl. Acad. Sci. USA 1942, 28, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Pampalona, J.; Frias, C.; Genesca, A.; Tusell, L. Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed]
- Lopez, V.; Barinova, N.; Onishi, M.; Pobiega, S.; Pringle, J.R.; Dubrana, K.; Marcand, S. Cytokinesis breaks dicentric chromosomes preferentially at pericentromeric regions and telomere fusions. Genes Dev. 2015, 29, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Hong, Y.; Sonneville, R.; Wang, B.; Scheidt, V.; Meier, B.; Woglar, A.; Demetriou, S.; Labib, K.; Jantsch, V.; Gartner, A. LEM-3 is a midbody-tethered DNA nuclease that resolves chromatin bridges during late mitosis. Nat. Commun. 2018, 9, 728. [Google Scholar] [CrossRef]
- Hong, Y.; Velkova, M.; Silva, N.; Jagut, M.; Scheidt, V.; Labib, K.; Jantsch, V.; Gartner, A. The conserved LEM-3/Ankle1 nuclease is involved in the combinatorial regulation of meiotic recombination repair and chromosome segregation in Caenorhabditis elegans. PLoS Genet. 2018, 14, e1007453. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001, 153, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Teitz, L.S.; Kim, D.H.; Shoshani, O.; Skaletsky, H.; Fachinetti, D.; Page, D.C.; Cleveland, D.W. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 2017, 19, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.; Korner, R.; Hofmann, K.; Nigg, E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 2007, 128, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Blackburn, E.H. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J. Cell Biol. 2004, 167, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, R.C.; de Boer, H.R.; Schoonen, P.M.; de Vries, E.G.; Lens, S.M.; van Vugt, M.A. Rif1 Is Required for Resolution of Ultrafine DNA Bridges in Anaphase to Ensure Genomic Stability. Dev. Cell 2015, 34, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Hickson, I.D. On the origins of ultra-fine anaphase bridges. Cell Cycle 2009, 8, 3065–3066. [Google Scholar] [CrossRef] [PubMed]
- Bizard, A.H.; Hickson, I.D. Anaphase: A fortune-teller of genomic instability. Curr. Opin. Cell Biol. 2018, 52, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; West, S.C. A new class of ultrafine anaphase bridges generated by homologous recombination. Cell Cycle 2018, 17, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.M.; Phua, H.H.; Mills, W.; Carpenter, A.J.; Porter, A.C.; Farr, C.J. Depletion of topoisomerase IIalpha leads to shortening of the metaphase interkinetochore distance and abnormal persistence of PICH-coated anaphase threads. J. Cell Sci. 2007, 120, 3952–3964. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Schwarzbraun, T.; Speicher, M.R.; Nigg, E.A. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma 2008, 117, 123–135. [Google Scholar] [CrossRef]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef]
- Waizenegger, I.C.; Hauf, S.; Meinke, A.; Peters, J.M. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 2000, 103, 399–410. [Google Scholar] [CrossRef]
- Haarhuis, J.H.; Elbatsh, A.M.; van den Broek, B.; Camps, D.; Erkan, H.; Jalink, K.; Medema, R.H.; Rowland, B.D. WAPL-mediated removal of cohesin protects against segregation errors and aneuploidy. Curr. Biol. 2013, 23, 2071–2077. [Google Scholar] [CrossRef]
- Piskadlo, E.; Tavares, A.; Oliveira, R.A. Metaphase chromosome structure is dynamically maintained by condensin I-directed DNA (de)catenation. eLife 2017, 6. [Google Scholar] [CrossRef]
- Hirano, T. Chromosome Dynamics during Mitosis. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Huttner, D.; Bizard, A.H.; Hirano, S.; Li, T.N.; Palmai-Pallag, T.; Bjerregaard, V.A.; Liu, Y.; Nigg, E.A.; Wang, L.H.; et al. PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat. Commun. 2015, 6, 8962. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Hickson, I.D. PICH promotes mitotic chromosome segregation: Identification of a novel role in rDNA disjunction. Cell Cycle 2016, 15, 2704–2711. [Google Scholar] [CrossRef] [PubMed]
- Kurasawa, Y.; Yu-Lee, L.Y. PICH and cotargeted Plk1 coordinately maintain prometaphase chromosome arm architecture. Mol. Biol. Cell 2010, 21, 1188–1199. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Addis Jones, O.; Chan, K.L. 53BP1 can limit sister-chromatid rupture and rearrangements driven by a distinct ultrafine DNA bridging-breakage process. Nat. Commun. 2018, 9, 677. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef]
- Barefield, C.; Karlseder, J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012, 40, 7358–7367. [Google Scholar] [CrossRef]
- Nera, B.; Huang, H.S.; Lai, T.; Xu, L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat. Commun. 2015, 6, 10132. [Google Scholar] [CrossRef] [PubMed]
- D’Alcontres, M.S.; Palacios, J.A.; Mejias, D.; Blasco, M.A. TopoIIalpha prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle 2014, 13, 1463–1481. [Google Scholar] [CrossRef] [PubMed]
- Hubner, N.C.; Wang, L.H.; Kaulich, M.; Descombes, P.; Poser, I.; Nigg, E.A. Re-examination of siRNA specificity questions role of PICH and Tao1 in the spindle checkpoint and identifies Mad2 as a sensitive target for small RNAs. Chromosoma 2010, 119, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Kaulich, M.; Cubizolles, F.; Nigg, E.A. On the regulation, function, and localization of the DNA-dependent ATPase PICH. Chromosoma 2012, 121, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Leng, M.; Besusso, D.; Jung, S.Y.; Wang, Y.; Qin, J. Targeting Plk1 to chromosome arms and regulating chromosome compaction by the PICH ATPase. Cell Cycle 2008, 7, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Albers, E.; Sbroggio, M.; Pladevall-Morera, D.; Bizard, A.H.; Avram, A.; Gonzalez, P.; Martin-Gonzalez, J.; Hickson, I.D.; Lopez-Contreras, A.J. Loss of PICH Results in Chromosomal Instability, p53 Activation, and Embryonic Lethality. Cell Rep. 2018, 24, 3274–3284. [Google Scholar] [CrossRef]
- Biebricher, A.; Hirano, S.; Enzlin, J.H.; Wiechens, N.; Streicher, W.W.; Huttner, D.; Wang, L.H.; Nigg, E.A.; Owen-Hughes, T.; Liu, Y.; et al. PICH: A DNA translocase specially adapted for processing anaphase bridge DNA. Mol. Cell 2013, 51, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Huh, J.W.; Warrington, R.; Li, B.; Wu, N.; Leng, M.; Zhang, J.; Ball, H.L.; Li, B.; Yu, H. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution. EMBO J. 2011, 30, 3309–3321. [Google Scholar] [CrossRef]
- Sarlos, K.; Biebricher, A.S.; Bizard, A.H.; Bakx, J.A.M.; Ferrete-Bonastre, A.G.; Modesti, M.; Paramasivam, M.; Yao, Q.; Peterman, E.J.G.; Wuite, G.J.L.; et al. Reconstitution of anaphase DNA bridge recognition and disjunction. Nat. Struct. Mol. Biol. 2018, 25, 868–876. [Google Scholar] [CrossRef]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef]
- Hickson, I.D. RecQ helicases: Caretakers of the genome. Nat. Rev. Cancer 2003, 3, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lonn, U.; Lonn, S.; Nylen, U.; Winblad, G.; German, J. An abnormal profile of DNA replication intermediates in Bloom’s syndrome. Cancer Res. 1990, 50, 3141–3145. [Google Scholar] [PubMed]
- Bachrati, C.Z.; Hickson, I.D. RecQ helicases: Guardian angels of the DNA replication fork. Chromosoma 2008, 117, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, R.S.; Schonberg, S.; German, J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 71, 4508–4512. [Google Scholar] [CrossRef] [PubMed]
- Bachrati, C.Z.; Hickson, I.D. RecQ helicases: Suppressors of tumorigenesis and premature aging. Biochem. J. 2003, 374, 577–606. [Google Scholar] [CrossRef] [PubMed]
- Rosin, M.P.; German, J. Evidence for chromosome instability in vivo in Bloom syndrome: Increased numbers of micronuclei in exfoliated cells. Hum. Genet. 1985, 71, 187–191. [Google Scholar] [CrossRef]
- Wu, L.; Davies, S.L.; North, P.S.; Goulaouic, H.; Riou, J.F.; Turley, H.; Gatter, K.C.; Hickson, I.D. The Bloom’s syndrome gene product interacts with topoisomerase III. J. Biol. Chem. 2000, 275, 9636–9644. [Google Scholar] [CrossRef]
- Hu, P.; Beresten, S.F.; van Brabant, A.J.; Ye, T.Z.; Pandolfi, P.P.; Johnson, F.B.; Guarente, L.; Ellis, N.A. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum. Mol. Genet. 2001, 10, 1287–1298. [Google Scholar] [CrossRef]
- Yin, J.; Sobeck, A.; Xu, C.; Meetei, A.R.; Hoatlin, M.; Li, L.; Wang, W. BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J. 2005, 24, 1465–1476. [Google Scholar] [CrossRef]
- Singh, T.R.; Ali, A.M.; Busygina, V.; Raynard, S.; Fan, Q.; Du, C.H.; Andreassen, P.R.; Sung, P.; Meetei, A.R. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008, 22, 2856–2868. [Google Scholar] [CrossRef]
- Xu, D.; Guo, R.; Sobeck, A.; Bachrati, C.Z.; Yang, J.; Enomoto, T.; Brown, G.W.; Hoatlin, M.E.; Hickson, I.D.; Wang, W. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 2008, 22, 2843–2855. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr.; Li, J.L.; Kenny, M.K.; Karow, J.K.; Cooper, M.P.; Kureekattil, R.P.; Hickson, I.D.; Bohr, V.A. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J. Biol. Chem. 2000, 275, 23500–23508. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.M.; Sommers, J.A.; Gray, M.D.; Lee, J.W.; von Kobbe, C.; Thoma, N.H.; Kureekattil, R.P.; Kenny, M.K.; Brosh, R.M., Jr. Physical and functional mapping of the replication protein a interaction domain of the werner and bloom syndrome helicases. J. Biol. Chem. 2005, 280, 29494–29505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [PubMed]
- Meetei, A.R.; Sechi, S.; Wallisch, M.; Yang, D.; Young, M.K.; Joenje, H.; Hoatlin, M.E.; Wang, W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 2003, 23, 3417–3426. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Wallis, J.W.; Chrebet, G.; Brodsky, G.; Rolfe, M.; Rothstein, R. A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 1989, 58, 409–419. [Google Scholar] [CrossRef]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef]
- Yang, J.; Bachrati, C.Z.; Ou, J.; Hickson, I.D.; Brown, G.W. Human topoisomerase IIIalpha is a single-stranded DNA decatenase that is stimulated by BLM and RMI1. J. Biol. Chem. 2010, 285, 21426–21436. [Google Scholar] [CrossRef]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase stimulates the activity of human topoisomerase IIIalpha. Nucleic Acids Res. 2002, 30, 4823–4829. [Google Scholar] [CrossRef]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Raynard, S.; Bussen, W.; Sung, P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J. Biol. Chem. 2006, 281, 13861–13864. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Singh, T.R.; Busygina, V.; Guo, R.; Wan, K.; Wang, W.; Sung, P.; Meetei, A.R.; Lei, M. Crystal structures of RMI1 and RMI2, two OB-fold regulatory subunits of the BLM complex. Structure 2010, 18, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.; Sarlos, K.; Logan, C.V.; Thakur, R.S.; Parry, D.A.; Bizard, A.H.; Leitch, A.; Cleal, L.; Ali, N.S.; Al-Owain, M.A.; et al. Mutations in TOP3A Cause a Bloom Syndrome-like Disorder. Am. J. Hum. Genet. 2018, 103, 456. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, P.; Godinho, S.A.; Parmar, K.; Pellman, D.; D’Andrea, A.D. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J. Clin. Investig. 2010, 120, 3834–3842. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.; Takai, H.; Buonomo, S.B.; Eisenhaber, F.; de Lange, T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004, 18, 2108–2119. [Google Scholar] [CrossRef]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hayano, M.; Masai, H. Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing. Trends Genet. 2013, 29, 449–460. [Google Scholar] [CrossRef]
- Xu, D.; Muniandy, P.; Leo, E.; Yin, J.; Thangavel, S.; Shen, X.; Ii, M.; Agama, K.; Guo, R.; Fox, D., 3rd; et al. Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 2010, 29, 3140–3155. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TOPBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Yamane, K.; Wu, X.; Chen, J. A DNA damage-regulated BRCT-containing protein, TOPBP1, is required for cell survival. Mol. Cell. Biol. 2002, 22, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Makiniemi, M.; Hillukkala, T.; Tuusa, J.; Reini, K.; Vaara, M.; Huang, D.; Pospiech, H.; Majuri, I.; Westerling, T.; Makela, T.P.; et al. BRCT domain-containing protein TOPBP1 functions in DNA replication and damage response. J. Biol. Chem. 2001, 276, 30399–30406. [Google Scholar] [CrossRef]
- Bang, S.W.; Ko, M.J.; Kang, S.; Kim, G.S.; Kang, D.; Lee, J.; Hwang, D.S. Human TOPBP1 localization to the mitotic centrosome mediates mitotic progression. Exp. Cell Res. 2011, 317, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TOPBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Germann, S.M.; Schramke, V.; Pedersen, R.T.; Gallina, I.; Eckert-Boulet, N.; Oestergaard, V.H.; Lisby, M. TOPBP1/DPB11 binds DNA anaphase bridges to prevent genome instability. J. Cell Biol. 2014, 204, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Broderick, R.; Nieminuszczy, J.; Blackford, A.N.; Winczura, A.; Niedzwiedz, W. TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat. Commun. 2015, 6, 6572. [Google Scholar] [CrossRef]
- Wardlaw, C.P.; Carr, A.M.; Oliver, A.W. TOPBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 2014, 22, 165–174. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The Causes and Consequences of Topological Stress during DNA Replication. Genes 2016, 7, 134. [Google Scholar] [CrossRef]
- Wyatt, H.D.; Laister, R.C.; Martin, S.R.; Arrowsmith, C.H.; West, S.C. The SMX DNA Repair Tri-nuclease. Mol. Cell 2017, 65, 848–860.e11. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Hirano, T. Dynamic organization of mitotic chromosomes. Curr. Opin. Cell Biol. 2017, 46, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Cuylen, S.; Metz, J.; Haering, C.H. Condensin structures chromosomal DNA through topological links. Nat. Struct. Mol. Biol. 2011, 18, 894–901. [Google Scholar] [CrossRef]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef]
- Ip, S.C.; Rass, U.; Blanco, M.G.; Flynn, H.R.; Skehel, J.M.; West, S.C. Identification of Holliday junction resolvases from humans and yeast. Nature 2008, 456, 357–361. [Google Scholar] [CrossRef]
- West, S.C.; Blanco, M.G.; Chan, Y.W.; Matos, J.; Sarbajna, S.; Wyatt, H.D. Resolution of Recombination Intermediates: Mechanisms and Regulation. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 103–109. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Casañas, M.; Chan, K.-L. The Unresolved Problem of DNA Bridging. Genes 2018, 9, 623. https://doi.org/10.3390/genes9120623
Fernández-Casañas M, Chan K-L. The Unresolved Problem of DNA Bridging. Genes. 2018; 9(12):623. https://doi.org/10.3390/genes9120623
Chicago/Turabian StyleFernández-Casañas, María, and Kok-Lung Chan. 2018. "The Unresolved Problem of DNA Bridging" Genes 9, no. 12: 623. https://doi.org/10.3390/genes9120623
APA StyleFernández-Casañas, M., & Chan, K.-L. (2018). The Unresolved Problem of DNA Bridging. Genes, 9(12), 623. https://doi.org/10.3390/genes9120623