Common Chromosomal Fragile Sites—Conserved Failure Stories


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Short Tandem DNA Repeats
3. Common Chromosomal Fragile Sites
4. Early Replicating Fragile Sites
5. Transcription-Replication Conflicts
6. Transcription Timing of Common Chromosomal Fragile Sites
7. Early Replicating Fragile Sites and Common Chromosomal Fragile Sites Comprise Underreplicated Regions in Mitosis
8. The Consequences of Underreplicated DNA
8.1. FANCD2 and Anaphase Bridges
8.2. Endonucleases Promote Common Chromosomal Fragile Sites Breakage and DNA Synthesis in Mitosis
8.3. Consequences of Underreplicated Regions in G1 Daughter Cells
9. Conserved Features of Common Chromosomal Fragile Sites Genes
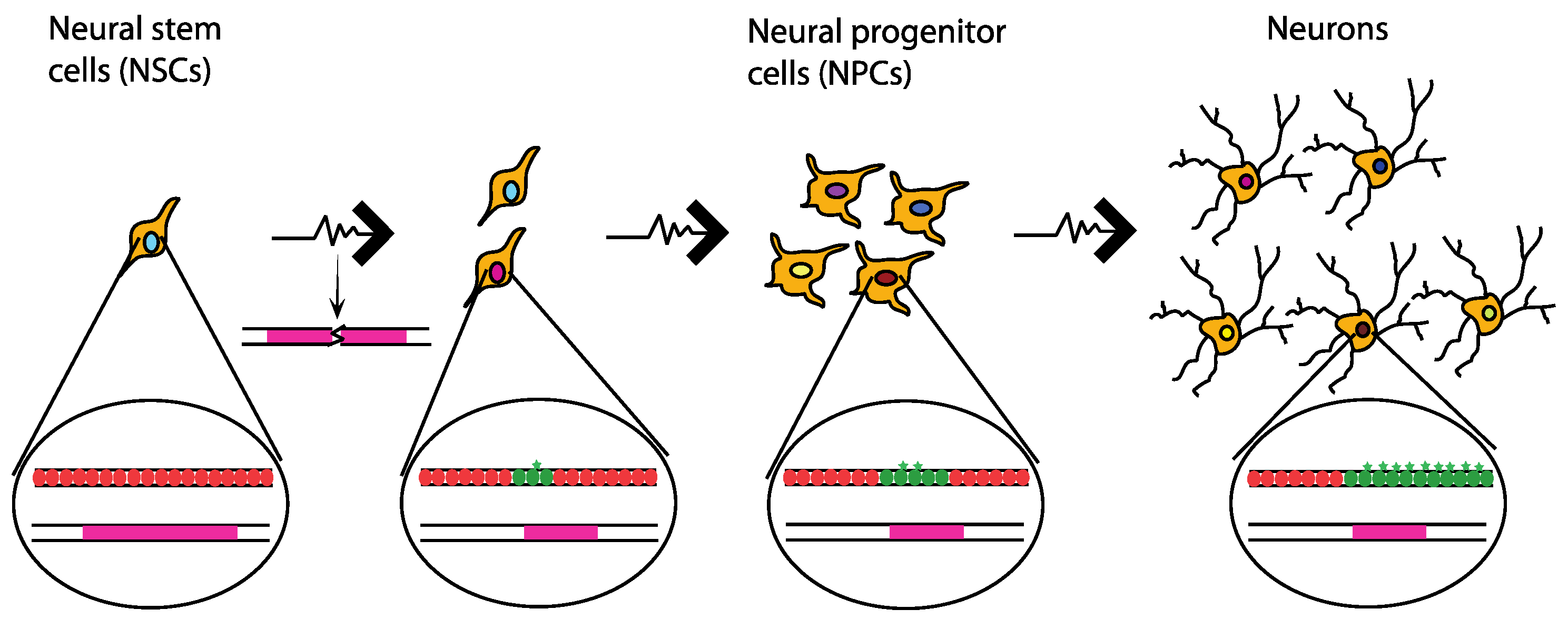
10. Neuronal Genetic Diversity Caused by Common Chromosomal Fragile Sites
11. A potential Link between Common Chromosomal Fragile Sites and LINE1-Dependent Neuronal Genetic Diversification
12. Potential Neuronal Epigenetic Diversity Caused by Common Chromosomal Fragile Sites
13. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kim, J.C.; Mirkin, S.M. The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 2013, 23, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Willems, T.; Guilmatre, A.; Zeng, H.; Markus, B.; Georgiev, S.; Daly, M.J.; Price, A.L.; Pritchard, J.K.; Sharp, A.J.; et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 2016, 48, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Quilez, J.; Guilmatre, A.; Garg, P.; Highnam, G.; Gymrek, M.; Erlich, Y.; Joshi, R.S.; Mittelman, D.; Sharp, A.J. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 2016, 44, 3750–3762. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.J. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 2018, 19, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Samadashwily, G.M.; Raca, G.; Mirkin, S.M. Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 1997, 17, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J.; Royle, N.J.; Wilson, V.; Wong, Z. Spontaneous mutation rates to new length alleles at tandem-repetitive hypervariable loci in human DNA. Nature 1988, 332, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Zoppis, S.; McCord, B. An overview of DNA typing methods for human identification: Past, present, and future. Methods Mol. Biol. 2012, 830, 3–16. [Google Scholar] [CrossRef]
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 2010, 11, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Hansen, R.S.; Canfield, T.K.; Lamb, M.M.; Gartler, S.M.; Laird, C.D. Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 1993, 73, 1403–1409. [Google Scholar] [CrossRef]
- Sutherland, G.R. Fragile sites on human chromosomes: Demonstration of their dependence on the type of tissue culture medium. Science 1977, 197, 265–266. [Google Scholar] [CrossRef]
- Pelletier, R.; Krasilnikova, M.M.; Samadashwily, G.M.; Lahue, R.; Mirkin, S.M. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 2003, 23, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef]
- Hecht, F.; Glover, T.W. Cancer chromosome breakpoints and common fragile sites induced by aphidicolin. Cancer Genet. Cytogenet. 1984, 13, 185–188. [Google Scholar] [CrossRef]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Dereli-Oz, A.; Versini, G.; Halazonetis, T.D. Studies of genomic copy number changes in human cancers reveal signatures of DNA replication stress. Mol. Oncol. 2011, 5, 308–314. [Google Scholar] [CrossRef]
- Karras, J.R.; Schrock, M.S.; Batar, B.; Huebner, K. Fragile genes that are frequently altered in cancer: Players not passengers. Cytogenet. Genome Res. 2016, 150, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 facilitates replication through common fragile sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Dutrillaux, B.; Lachages, A.M.; Millot, G.A.; Brison, O.; Debatisse, M. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 2011, 18, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Pentzold, C.; Shah, S.A.; Hansen, N.R.; Le Tallec, B.; Seguin-Orlando, A.; Debatisse, M.; Lisby, M.; Oestergaard, V.H. FANCD2 binding identifies conserved fragile sites at large transcribed genes in avian cells. Nucleic Acids Res. 2018, 46, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Aguilera, A. Transcription as a threat to genome integrity. Annu. Rev. Biochem. 2016, 85, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Oestergaard, V.H.; Lisby, M. Transcription-replication conflicts at chromosomal fragile sites-consequences in M phase and beyond. Chromosoma 2017, 126, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Gros, J.; Kumar, C.; Lynch, G.; Yadav, T.; Whitehouse, I.; Remus, D. Post-licensing specification of eukaryotic replication origins by facilitated Mcm2-7 sliding along DNA. Mol. Cell 2015, 60, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Freudenreich, C.H. R-loops: Targets for nuclease cleavage and repeat instability. Curr. Genet. 2018, 64, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Su, X.A.; Freudenreich, C.H. Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, E8392–E8401. [Google Scholar] [CrossRef] [PubMed]
- Neil, A.J.; Liang, M.U.; Khristich, A.N.; Shah, K.A.; Mirkin, S.M. RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res. 2018, 46, 3487–3497. [Google Scholar] [CrossRef] [PubMed]
- Meryet-Figuiere, M.; Alaei-Mahabadi, B.; Ali, M.M.; Mitra, S.; Subhash, S.; Pandey, G.K.; Larsson, E.; Kanduri, C. Temporal separation of replication and transcription during S-phase progression. Cell Cycle 2014, 13, 3241–3248. [Google Scholar] [CrossRef]
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1506. [Google Scholar] [CrossRef]
- Schubeler, D.; Scalzo, D.; Kooperberg, C.; van Steensel, B.; Delrow, J.; Groudine, M. Genome-wide DNA replication profile for Drosophila melanogaster: A link between transcription and replication timing. Nat. Genet. 2002, 32, 438–442. [Google Scholar] [CrossRef]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R., 3rd; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef]
- Techer, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachages, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol. Cell 2018, 71, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M.; Lowndes, N.F. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Syljuasen, R.G.; Lukas, J.; Bartek, J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle 2004, 3, 941–945. [Google Scholar] [CrossRef]
- Torres-Rosell, J.; De Piccoli, G.; Cordon-Preciado, V.; Farmer, S.; Jarmuz, A.; Machin, F.; Pasero, P.; Lisby, M.; Haber, J.E.; Aragon, L. Anaphase onset before complete DNA replication with intact checkpoint responses. Science 2007, 315, 1411–1415. [Google Scholar] [CrossRef]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef]
- Bhat, A.; Andersen, P.L.; Qin, Z.; Xiao, W. Rev3, the catalytic subunit of Polζ, is required for maintaining fragile site stability in human cells. Nucleic Acids Res. 2013, 41, 2328–2339. [Google Scholar] [CrossRef]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Gallina, I.; Christiansen, S.K.; Pedersen, R.T.; Lisby, M.; Oestergaard, V.H. TopBP1-mediated DNA processing during mitosis. Cell Cycle 2016, 15, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017, 31, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Iwasaki, W.M.; Kugou, K.; Takahashi, K.K.; Oda, A.; Sato, K.; Kobayashi, W.; Kawai, H.; Sakasai, R.; Takaori-Kondo, A.; et al. Replication stress induces accumulation of FANCD2 at central region of large fragile genes. Nucleic Acids Res. 2018, 46, 2932–2944. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Arlt, M.F.; Howlett, N.G.; Glover, T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006, 25, 4381–4388. [Google Scholar] [CrossRef]
- Arlt, M.F.; Xu, B.; Durkin, S.G.; Casper, A.M.; Kastan, M.B.; Glover, T.W. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol. Cell. Biol. 2004, 24, 6701–6709. [Google Scholar] [CrossRef]
- Chan, K.L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Ira, G. Break-induced replication: Functions and molecular mechanism. Curr. Opin. Genet. Dev. 2013, 23, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.P.; Lovett, S.T.; Haber, J.E. Break-induced DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010397. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Huhn, D.; et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol. Cell 2017, 66, 658–671.e8. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, H.D.; Sarbajna, S.; Matos, J.; West, S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 2013, 52, 234–247. [Google Scholar] [CrossRef]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef]
- Germann, S.M.; Schramke, V.; Pedersen, R.T.; Gallina, I.; Eckert-Boulet, N.; Oestergaard, V.H.; Lisby, M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J. Cell Biol. 2014, 204, 45–59. [Google Scholar] [CrossRef]
- Gritenaite, D.; Princz, L.N.; Szakal, B.; Bantele, S.C.; Wendeler, L.; Schilbach, S.; Habermann, B.H.; Matos, J.; Lisby, M.; Branzei, D.; et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014, 28, 1604–1619. [Google Scholar] [CrossRef]
- Deng, L.; Wu, R.A.; Kochenova, O.V.; Pellman, D.S.; Walter, J.C. Mitotic CDK promotes replisome disassembly, fork breakage, and complex DNA rearrangements. BioRxiv 2018. [Google Scholar] [CrossRef]
- Glover, T.W.; Stein, C.K. Induction of sister chromatid exchanges at common fragile sites. Am. J. Hum. Genet. 1987, 41, 882–890. [Google Scholar] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Vignard, J.; Mirey, G. Around and beyond 53BP1 Nuclear Bodies. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Lezaja, A.; Altmeyer, M. Inherited DNA lesions determine G1 duration in the next cell cycle. Cell Cycle 2018, 17, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novak, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 14728. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Moser, J.; Phadke, H.; Basha, A.A.; Spencer, S.L. Endogenous replication stress in mother cells leads to quiescence of daughter cells. Cell Rep. 2017, 19, 1351–1364. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Bonnet, A.; Grosso, A.R.; Elkaoutari, A.; Coleno, E.; Presle, A.; Sridhara, S.C.; Janbon, G.; Geli, V.; de Almeida, S.F.; Palancade, B. Introns protect eukaryotic genomes from transcription-associated genetic instability. Mol. Cell 2017, 67, 608–621.e6. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed]
- Manzo, S.G.; Hartono, S.R.; Sanz, L.A.; Marinello, J.; De Biasi, S.; Cossarizza, A.; Capranico, G.; Chedin, F. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 2018, 19, 100. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.A.; Gregory, T.R.; Witt, C.C. Metabolic ‘engines’ of flight drive genome size reduction in birds. Proc. Biol. Sci. 2014, 281, 20132780. [Google Scholar] [CrossRef]
- Wei, P.C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long neural genes harbor recurrent dna break clusters in neural stem/progenitor cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E. Molecular biology: Breaks in the brain. Nature 2016, 532, 46–47. [Google Scholar] [CrossRef]
- Finlay, B.L.; Slattery, M. Local differences in the amount of early cell death in neocortex predict adult local specializations. Science 1983, 219, 1349–1351. [Google Scholar] [CrossRef]
- Ferrer, I.; Soriano, E.; del Rio, J.A.; Alcantara, S.; Auladell, C. Cell death and removal in the cerebral cortex during development. Prog. Neurobiol. 1992, 39, 1–43. [Google Scholar] [CrossRef]
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014, 8, 1280–1289. [Google Scholar] [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Katyal, S.; Downing, S.M.; Zhao, J.; Russell, H.R.; McKinnon, P.J. Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat. Neurosci. 2012, 15, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Wang, S.; Jiang, L.; Kang, J.; Benraiss, A.; Harrison-Restelli, C.; Fraser, R.A.; Couldwell, W.T.; Kawaguchi, A.; Okano, H.; et al. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat. Med. 2000, 6, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G., 2nd; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Bostrom, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Han, J.; Cheng, E.C.; Yamaguchi, S.; Shima, N.; Thomas, J.L.; Lin, H. Embryonic stem cells license a high level of dormant origins to protect the genome against replication stress. Stem Cell Rep. 2015, 5, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Erwin, J.A.; Marchetto, M.C.; Gage, F.H. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 2014, 15, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Singer, T.; McConnell, M.J.; Marchetto, M.C.; Coufal, N.G.; Gage, F.H. LINE-1 retrotransposons: Mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010, 33, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.; Lutz, S.; Morrish, T.A.; Moran, J.V. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol. Cell. Biol. 2005, 25, 7780–7795. [Google Scholar] [CrossRef] [PubMed]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef]
- Kim, J.; Sturgill, D.; Sebastian, R.; Khurana, S.; Tran, A.D.; Edwards, G.B.; Kruswick, A.; Burkett, S.; Hosogane, E.K.; Hannon, W.W.; et al. Replication stress shapes a protective chromatin environment across fragile genomic regions. Mol. Cell 2018, 69, 36–47.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Guilbaud, G.; Schiavone, D.; Sale, J.E. Nucleotide pool depletion induces g-quadruplex-dependent perturbation of gene expression. Cell Rep. 2015, 13, 2491–2503. [Google Scholar] [CrossRef]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voutsinos, V.; Munk, S.H.N.; Oestergaard, V.H. Common Chromosomal Fragile Sites—Conserved Failure Stories. Genes 2018, 9, 580. https://doi.org/10.3390/genes9120580
Voutsinos V, Munk SHN, Oestergaard VH. Common Chromosomal Fragile Sites—Conserved Failure Stories. Genes. 2018; 9(12):580. https://doi.org/10.3390/genes9120580
Chicago/Turabian StyleVoutsinos, Vasileios, Sebastian H. N. Munk, and Vibe H. Oestergaard. 2018. "Common Chromosomal Fragile Sites—Conserved Failure Stories" Genes 9, no. 12: 580. https://doi.org/10.3390/genes9120580
APA StyleVoutsinos, V., Munk, S. H. N., & Oestergaard, V. H. (2018). Common Chromosomal Fragile Sites—Conserved Failure Stories. Genes, 9(12), 580. https://doi.org/10.3390/genes9120580