WebCircRNA: Classifying the Circular RNA Potential of Coding and Noncoding RNA

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of Datasets
2.2. Feature Encoding
2.3. Random Forest Models
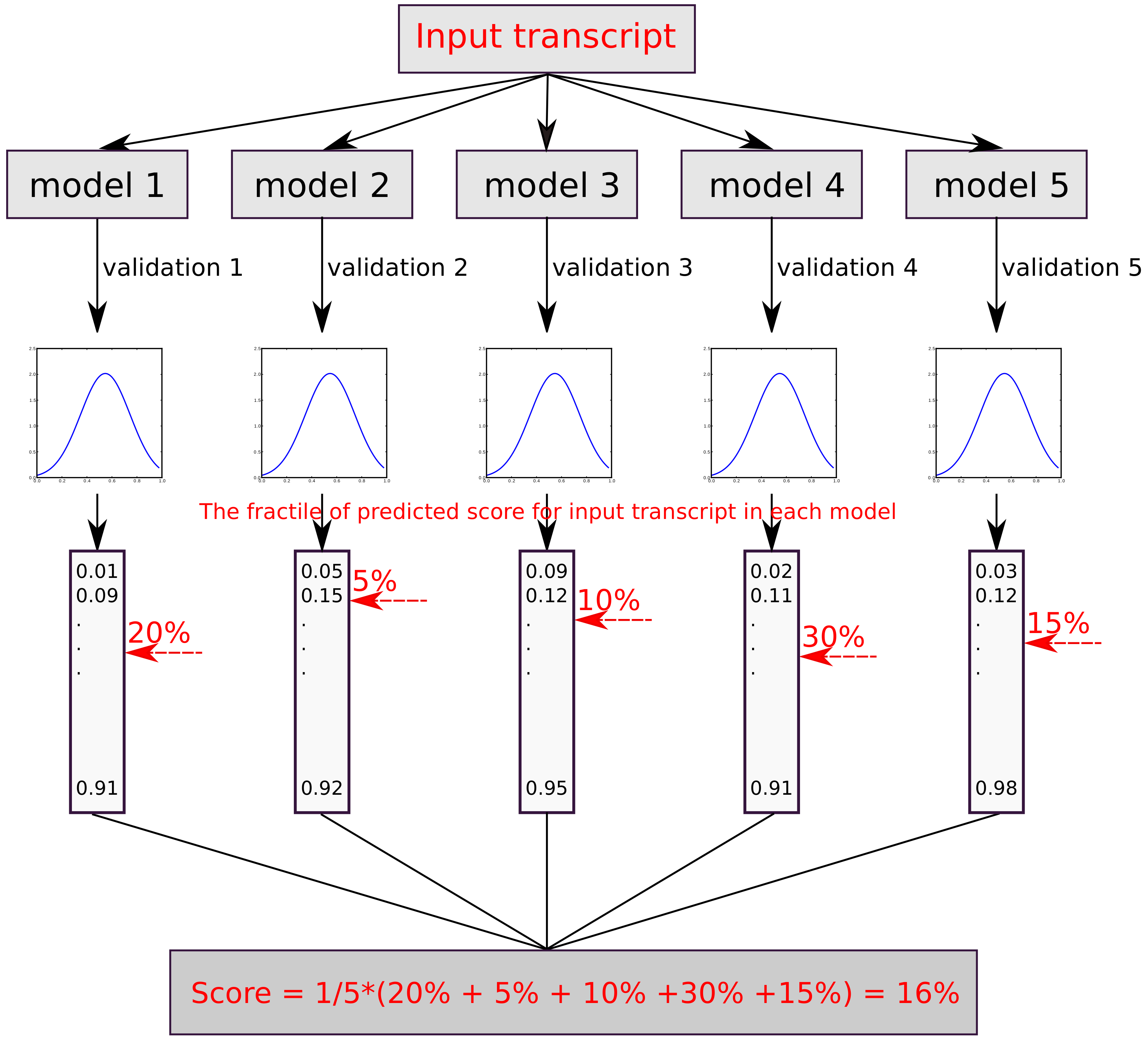
2.4. Prediction Scores
3. Results
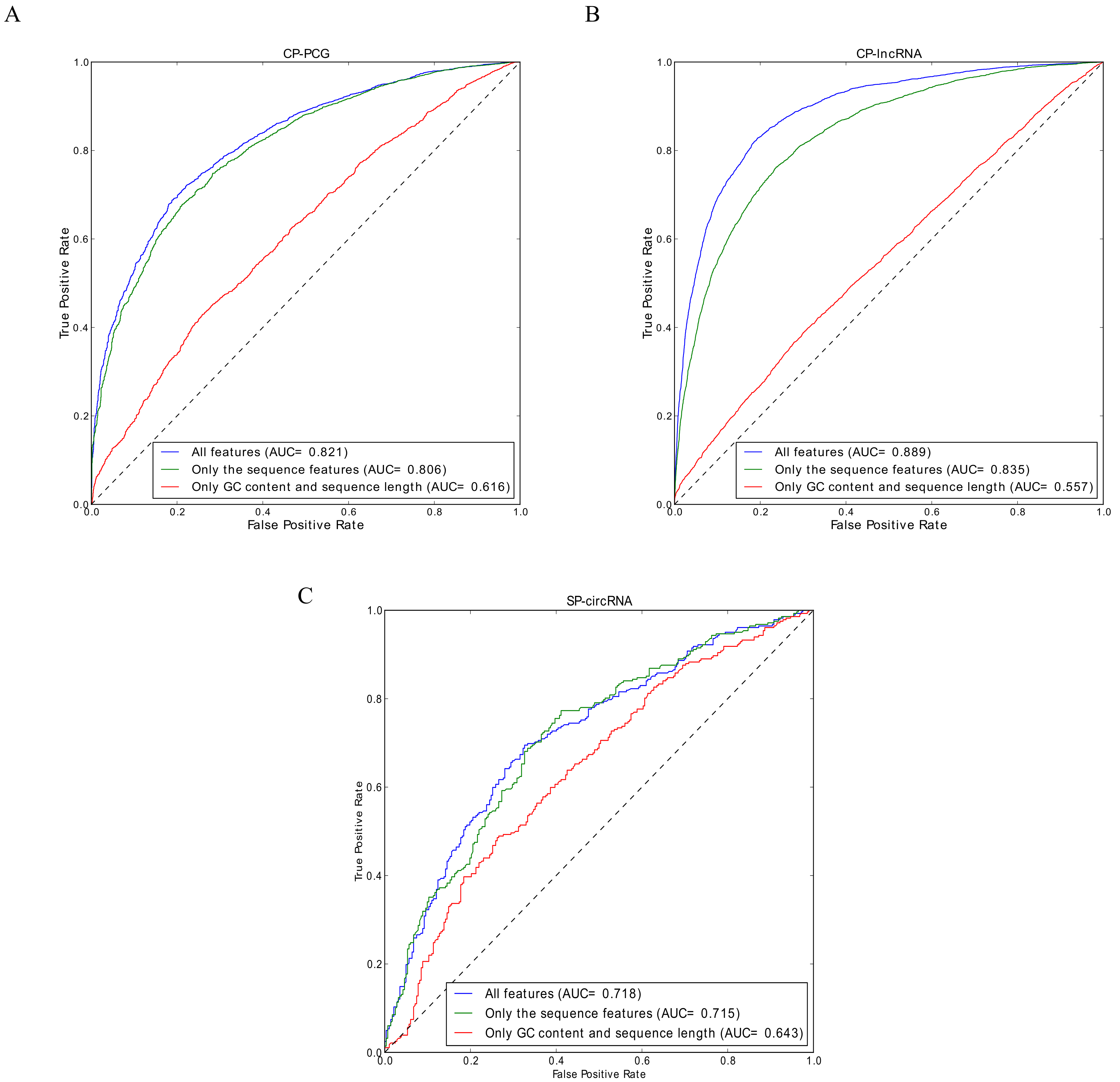
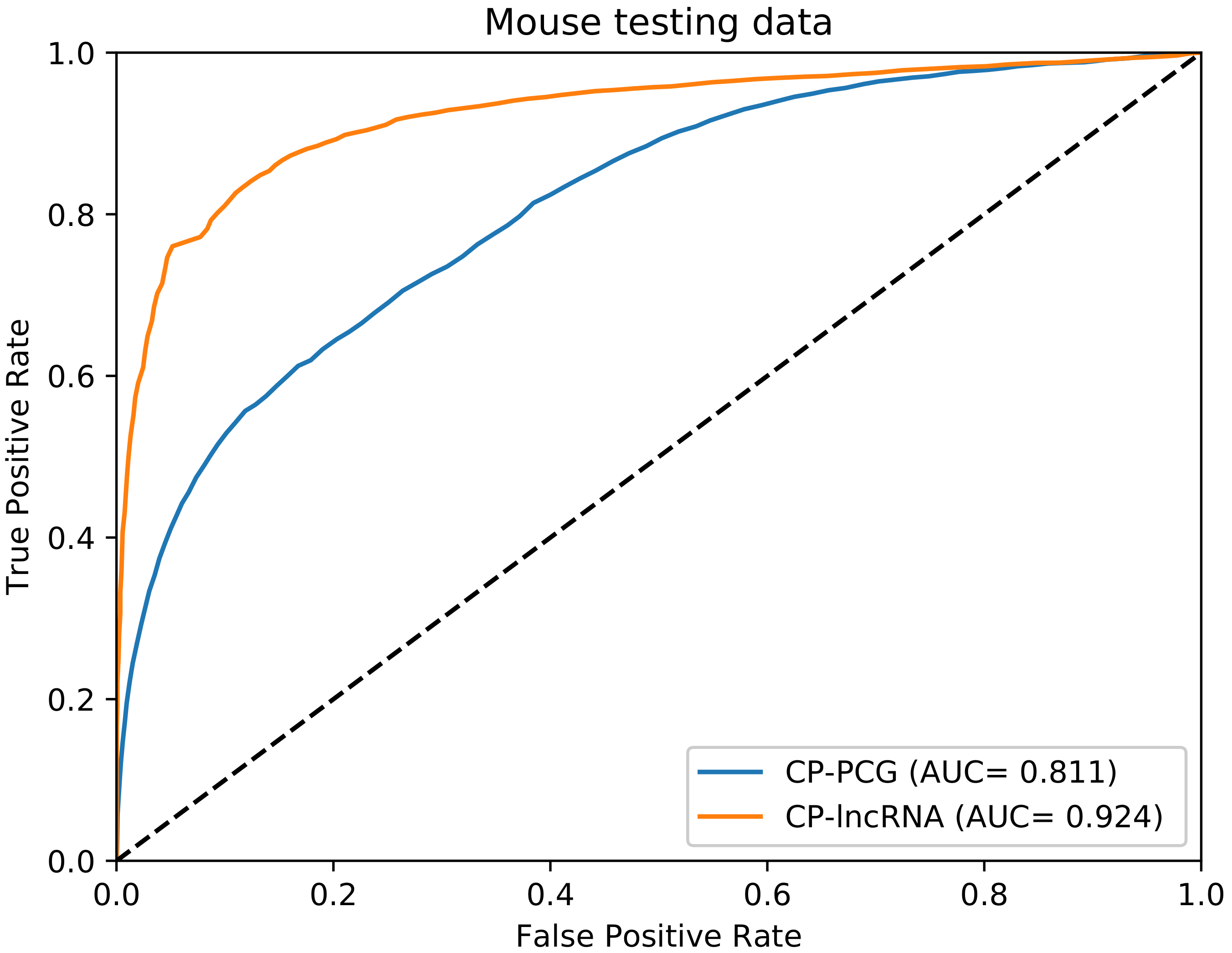
3.1. Performance Evaluation
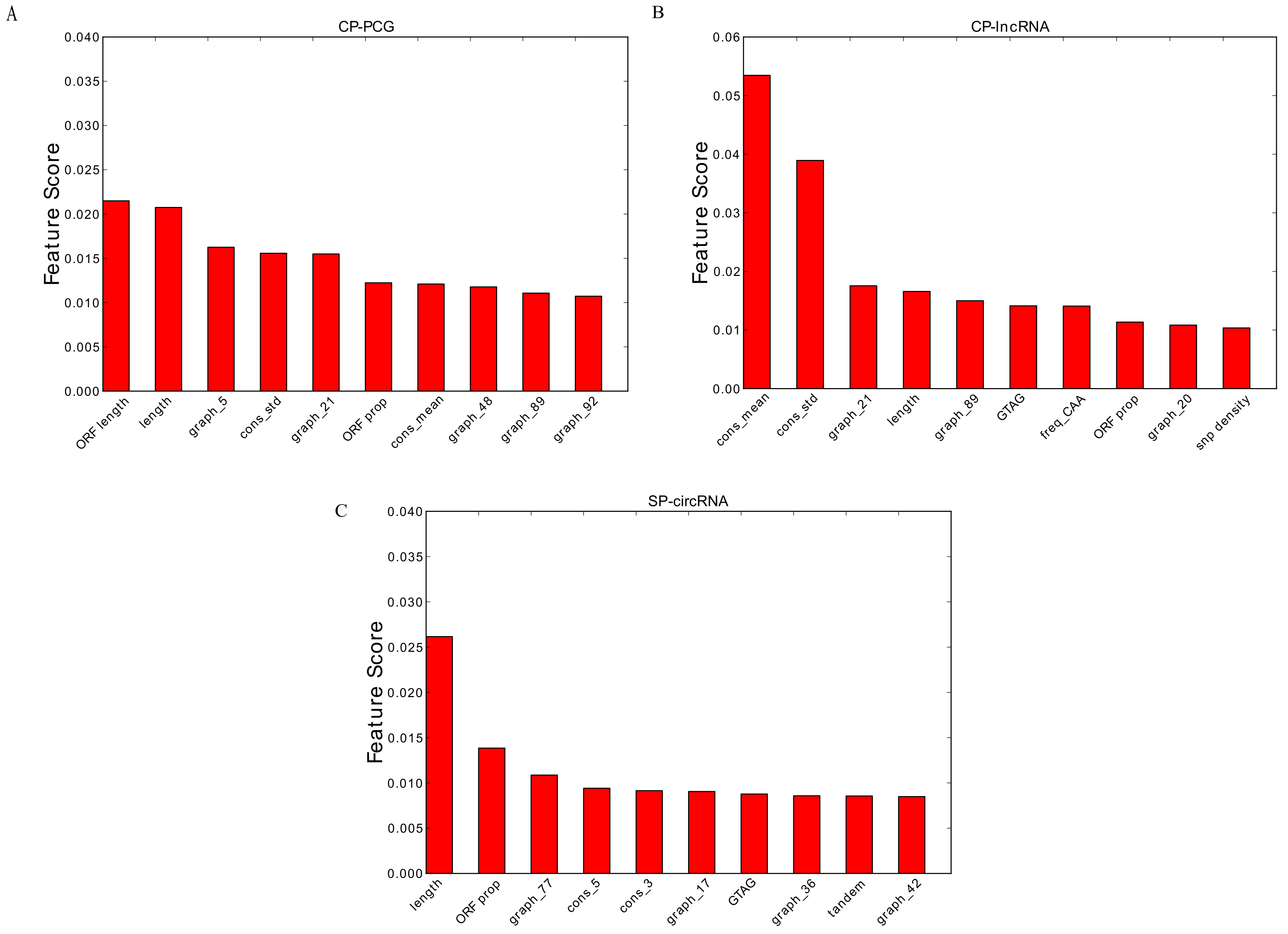
3.2. Feature Importance
3.3. The WebCircRNA Web Server
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| circRNA | circular RNA |
| lncRNA | long non-coding RNA |
| PCG | protein coding gene |
| ROC | receiver operating characteristic |
| AUC | the area under the ROC curve |
| ORF | open reading frame |
| FPR | false positive rate |
| CP-lncRNA | circRNA potential of lncRNA |
| CP-PCG | circRNA potential of PCG |
| CP-lncRNA | circRNA potential of lncRNA |
| SP-circRNA | stem cell potential of circRNA |
References
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, Y.; Zheng, Y.; Huang, Y.; Zhang, Y.; Jia, L.; Li, W. Circular RNA CDR1as regulates osteoblastic differentiation of periodontal ligament stem cells via the miR-7/GDF5/SMAD and p38 MAPK signaling pathway. Stem Cell Res. Ther. 2018, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Matsumoto, K.; Nishihara, M.; Nakano, Y.; Shibata, A.; Maruyama, H.; Shuto, S.; Matsuda, A.; Yoshida, M.; Ito, Y.; et al. Rolling circle translation of circular RNA in living human cells. Sci. Rep. 2015, 5, 16435. [Google Scholar] [CrossRef] [PubMed]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. CircBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Venø, M.T.; Hansen, T.B.; Venø, S.T.; Clausen, B.H.; Grebing, M.; Finsen, B.; Holm, I.E.; Kjems, J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015, 16, 245. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Venø, M.T.; Damgaard, C.K.; Kjems, J. Comparison of circular RNA prediction tools. Nucleic Acids Res. 2016, 44, e58. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Xiong, K. PredcircRNA: Computational classification of circular RNA from other long non-coding RNA using hybrid features. Mol. Biosyst. 2015, 11, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Kitamura-Abe, S.; Itoh, H.; Washio, T.; Tsutsumi, A.; Tomita, M. Characterization of the splice sites in GT-AG and GC-AG introns. J. Bioinform. Comput. Biol. 2004, 2, 309–331. [Google Scholar] [CrossRef] [PubMed]
- Maticzka, D.; Lange, S.J.; Costa, F.; Backofen, R. GraphProt: Modeling binding preferences of RNA-binding proteins. Genome Biol. 2014, 15, R17. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Lowe, C.B.; Kellis, M.; Siepel, A.; Raney, B.J.; Clamp, M.; Salama, S.R.; Kingsley, D.M.; Lindblad-Toh, K.; Haussler, D. Three periods of regulatory innovation during vertebrate evolution. Science 2011, 333, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Speir, M.L.; Zweig, A.S.; Rosenbloom, K.R.; Raney, B.J.; Paten, B.; Nejad, P.; Lee, B.T.; Learned, K.; Karolchik, D.; Hinrichs, A.S. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016, 44, D717–D725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cowled, C.; Shi, Z.; Huang, Z.; Bishop-Lilly, K.A.; Fang, X.; Wynne, J.W.; Xiong, Z.; Baker, M.L.; Zhao, W. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 2013, 339, 456–460. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012, 28, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010, 6, e1001223. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef]
- Manuel, F.; Cernadas, E.; Barro, S.; Amorim, D. Do we need hundreds of classifiers to solve real world classification problems? J. Mach. Learn. Res. 2014, 15, 3133–3181. [Google Scholar]
Sample Availability: The web server, datasets and standalone python code supporting the findings of this study are available at http://rth.dk/resources/webcircrna. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Positive Data | Negative Data |
|---|---|---|
| circRNA vs. PCG | Total: 14,084 circRNAs | Total: 9533 PCGs not overlapping with circRNAs |
| Training: 10,000 | Training: 8000 | |
| Independent testing: 4084 | Independent testing: 1533 | |
| circRNA vs. lncRNA | Total: 14,084 circRNAs | Total: 19,722 lncRNAs not overlapping with circRNAs |
| Training: 10,000 | Training: 10,000 | |
| Independent testing: 4084 | Independent testing: 9722 | |
| Stem cell vs. not | Total: 2082 circRNAs | Total: 2082 circRNAs |
| Training: 1800 | Training: 1800 | |
| Independent testing: 282 | Independent testing: 282 |
| Feature Group | Feature Names |
|---|---|
| Basic sequence features | Length; AG, GT, GTAG, AGGT, GC content; 64 trinucleotide frequencies |
| Graph features | Top 101 graph features from GraphProt 1.0.1 |
| Conservation features | Mean, standard deviation of conservation score |
| Other features | ALU, tandem, ORF length, ORF prop, SNP density |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Xiong, K.; Anthon, C.; Hyttel, P.; Freude, K.K.; Jensen, L.J.; Gorodkin, J. WebCircRNA: Classifying the Circular RNA Potential of Coding and Noncoding RNA. Genes 2018, 9, 536. https://doi.org/10.3390/genes9110536
Pan X, Xiong K, Anthon C, Hyttel P, Freude KK, Jensen LJ, Gorodkin J. WebCircRNA: Classifying the Circular RNA Potential of Coding and Noncoding RNA. Genes. 2018; 9(11):536. https://doi.org/10.3390/genes9110536
Chicago/Turabian StylePan, Xiaoyong, Kai Xiong, Christian Anthon, Poul Hyttel, Kristine K. Freude, Lars Juhl Jensen, and Jan Gorodkin. 2018. "WebCircRNA: Classifying the Circular RNA Potential of Coding and Noncoding RNA" Genes 9, no. 11: 536. https://doi.org/10.3390/genes9110536
APA StylePan, X., Xiong, K., Anthon, C., Hyttel, P., Freude, K. K., Jensen, L. J., & Gorodkin, J. (2018). WebCircRNA: Classifying the Circular RNA Potential of Coding and Noncoding RNA. Genes, 9(11), 536. https://doi.org/10.3390/genes9110536