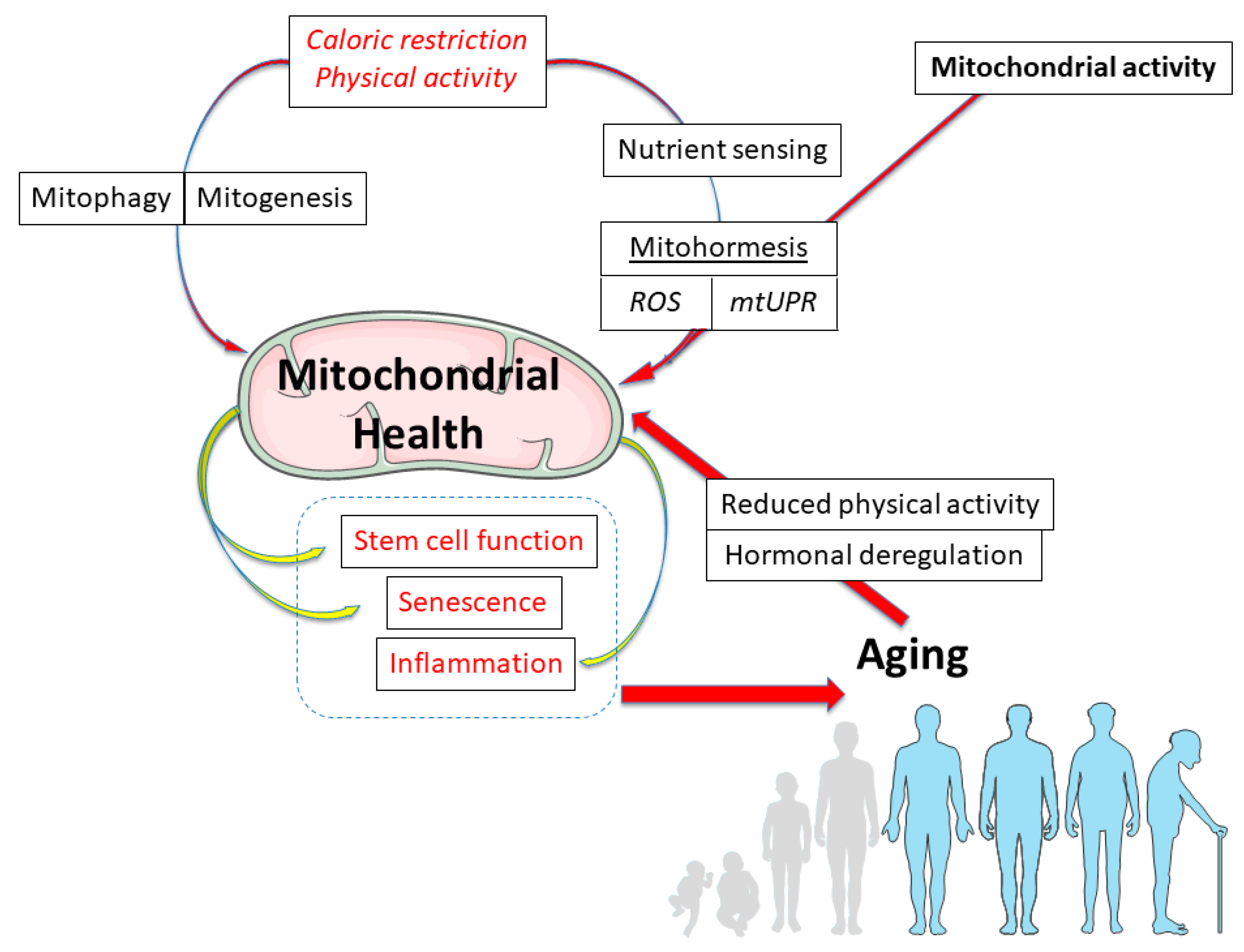
The Aging Mitochondria



{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Dysfunction Is Associated with Aging
3. From the Mitochondrial Free Radical Theory of Aging to a More Complex View
4. A Complex and Mitochondria-Centered View of Cell Dysfunctions in Aging
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hughes, K.A.; Reynolds, R.M. Evolutionary and mechanistic theories of aging. Annu. Rev. Entomol. 2005, 50, 421–445. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Invest. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria and ageing: Role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; Dantona, G.; Nisoli, E.; Flati, V.; Assanelli, D.; Dioguardi, F.S.; Bianchi, R. Morphometric changes induced by amino acid supplementation in skeletal and cardiac muscles of old mice. Am. J. Cardiol. 2008, 101, 26E–34E. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Sato, T. Age changes in size and number of mitochondria of human hepatic cells. J. Gerontol. 1968, 23, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Herbener, G.H. A morphometric study of age-dependent changes in mitochondrial population of mouse liver and heart. J. Gerontol. 1976, 31, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ulloa, A.; Nogueira, L.; Rodriguez, A.; Barboza, J.; Hogan, M.C.; Ceballos, G.; Villarreal, F.; Ramirez-Sanchez, I. Recovery of Indicators of Mitochondrial Biogenesis, Oxidative Stress, and Aging With (-)-Epicatechin in Senile Mice. J. Gerontol. A 2015, 70, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.M.; Hood, D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Oberlin, A.S.; Holmuhamedov, E.L.; Gupta, A.; Sagar, S.; Syed, R.H.; Siddiqui, S.A.; Raghavakaimal, S.; Terzic, A.; Jahangir, A. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech. Ageing Dev. 2008, 129, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Ojaimi, J.; Masters, C.L.; Opeskin, K.; McKelvie, P.; Byrne, E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 1999, 111, 39–47. [Google Scholar] [CrossRef]
- Sastre, J.; Pallardo, F.V.; Pla, R.; Pellin, A.; Juan, G.; O’Connor, J.E.; Estrela, J.M.; Miquel, J.; Vina, J. Aging of the liver: Age-associated mitochondrial damage in intact hepatocytes. Hepatology 1996, 24, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.C.; Su, J.H.; King, K.L.; Wei, Y.H. Ageing-associated 5 kb deletion in human liver mitochondrial DNA. Biochem. Biophys. Res. Commun. 1991, 178, 124–131. [Google Scholar] [CrossRef]
- Fayet, G.; Jansson, M.; Sternberg, D.; Moslemi, A.R.; Blondy, P.; Lombes, A.; Fardeau, M.; Oldfors, A. Ageing muscle: Clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul. Disord. 2002, 12, 484–493. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef]
- Mecocci, P.; Fano, G.; Fulle, S.; MacGarvey, U.; Shinobu, L.; Polidori, M.C.; Cherubini, A.; Vecchiet, J.; Senin, U.; Beal, M.F. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 1999, 26, 303–308. [Google Scholar] [CrossRef]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Heath, S.H.; Hagen, T.M. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic. Biol. Med. 2003, 35, 1064–1072. [Google Scholar] [CrossRef]
- Choksi, K.B.; Nuss, J.E.; Deford, J.H.; Papaconstantinou, J. Age-related alterations in oxidatively damaged proteins of mouse skeletal muscle mitochondrial electron transport chain complexes. Free Radic. Biol. Med. 2008, 45, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Packer, L.; Fuehr, K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 1977, 267, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; von Zglinicki, T.; Lorenz, M.; Saretzki, G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J. Biol. Chem. 2003, 278, 6824–6830. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; de Oliveira, R.M.; Conboy, C.M.; Haigis, M.; Guarente, L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem. 2003, 278, 38966–38969. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Cao, P.; Thiels, E.; Chu, C.T.; Wu, G.Y.; Oury, T.D.; Klann, E. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol. Learn. Mem. 2007, 87, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.D.; Christopher, N.C.; Lateef, S.; Elamir, B.M.; Patel, M.; Liang, L.P.; Crapo, J.D. Extracellular superoxide dismutase overexpression protects against aging-induced cognitive impairment in mice. Behav. Genet. 2002, 32, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.D.; Christopher, N.C.; Crapo, J.D. Memory decline of aging reduced by extracellular superoxide dismutase overexpression. Behav. Genet. 2005, 35, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.G.; Callahan, D.M.; Foulis, S.A.; Kent-Braun, J.A. Age-related changes in oxidative capacity differ between locomotory muscles and are associated with physical activity behavior. Appl. Physiol. Nutr. Metab. 2012, 37, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Begin, M.E.; Langlois, M.F.; Lorrain, D.; Cunnane, S.C. Thyroid Function and Cognition during Aging. Curr. Gerontol. Geriatr. Res. 2008, 2008, 474868. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.M.; Iwen, K.A.; Seitz, H.J. Regulation of mitochondrial biogenesis by thyroid hormone. Exp. Physiol. 2003, 88, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.A.; Maddalena, L.A.; Merilovich, M.; Robb, E.L. A midlife crisis for the mitochondrial free radical theory of aging. Longev. Healthspan 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Hekimi, S. When a theory of aging ages badly. Cell. Mol. Life Sci. 2010, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.G.; Trifunovic, A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009, 10, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, L.A.; Tooley, J.G.; Van Hoose, P.M.; Wang, E.; Cheng, A.; Cole, M.P.; Schaner Tooley, C.E. NRMT1 knockout mice exhibit phenotypes associated with impaired DNA repair and premature aging. Mech. Ageing Dev. 2015, 146–148, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Perez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. J. Gerontol. A 2009, 64, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Beadle, N.E.; Taylor, G.A.; Commane, D.; Mathers, J.C.; Khrapko, K.; Turnbull, D.M. Quantification of mitochondrial DNA mutation load. Aging Cell 2009, 8, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Schon, E.A.; Chinnery, P.F. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004, 20, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.M.; Stewart, J.B.; Hagstrom, E.; Brene, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Vijg, J. Mitochondrial DNA mutations and aging: A case closed? Nat. Genet. 2007, 39, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Kraytsberg, Y.; de Grey, A.D.; Vijg, J.; Schon, E.A. Does premature aging of the mtDNA mutator mouse prove that mtDNA mutations are involved in natural aging? Aging Cell 2006, 5, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Bielas, J.H.; Kujoth, G.C.; Ladiges, W.C.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007, 39, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Huang, J.; Edwards, Y.J.; Ulloa, R.H.; Dillon, L.M.; Prolla, T.A.; Vance, J.M.; Moraes, C.T.; Zuchner, S. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010, 12, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, J.; Ohta, S.; Kikuchi, A.; Takemitsu, M.; Goto, Y.; Nonaka, I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 1991, 88, 10614–10618. [Google Scholar] [CrossRef] [PubMed]
- Holme, E.; Larsson, N.G.; Oldfors, A.; Tulinius, M.; Sahlin, P.; Stenman, G. Multiple symmetric lipomas with high levels of mtDNA with the tRNA(Lys) A—>G(8344) mutation as the only manifestation of disease in a carrier of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am. J. Hum. Genet. 1993, 52, 551–556. [Google Scholar] [PubMed]
- Chomyn, A.; Martinuzzi, A.; Yoneda, M.; Daga, A.; Hurko, O.; Johns, D.; Lai, S.T.; Nonaka, I.; Angelini, C.; Attardi, G. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 1992, 89, 4221–4225. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Samuels, D.C. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am. J. Hum. Genet. 1999, 64, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Durham, S.E.; Bonilla, E.; Samuels, D.C.; DiMauro, S.; Chinnery, P.F. Mitochondrial DNA copy number threshold in mtDNA depletion myopathy. Neurology 2005, 65, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hamalainen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Gotz, A.; Forsstrom, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Rando, T.A. Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle 2012, 11, 2260–2267. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.E.; Conboy, M.J.; Hsu, M.; Barchas, L.; Jeong, J.; Agrawal, A.; Mikels, A.J.; Agrawal, S.; Schaffer, D.V.; Conboy, I.M. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell 2009, 8, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Hamalainen, R.H.; Ahlqvist, K.J.; Ellonen, P.; Lepisto, M.; Logan, A.; Otonkoski, T.; Murphy, M.P.; Suomalainen, A. mtDNA Mutagenesis Disrupts Pluripotent Stem Cell Function by Altering Redox Signaling. Cell Rep. 2015, 11, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014, 33, 1321–1340. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Dohla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Easlon, E.; Tsang, F.; Skinner, C.; Wang, C.; Lin, S.J. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008, 22, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.A.; Delaney, J.R.; Tsuchiya, M.; Tsuchiyama, S.; Shemorry, A.; Sim, S.; Chou, A.C.; Ahmed, U.; Carr, D.; Murakami, C.J.; et al. A Comprehensive Analysis of Replicative Lifespan in 4,698 Single-Gene Deletion Strains Uncovers Conserved Mechanisms of Aging. Cell Metab. 2015, 22, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.B.; Copes, N.; Brito, A.G.; Canfield, J.; Bradshaw, P.C. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS ONE 2013, 8, e58345. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Choi, K.M.; Lee, C.K. Caloric restriction improves efficiency and capacity of the mitochondrial electron transport chain in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2011, 409, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Bonawitz, N.D.; Chatenay-Lapointe, M.; Pan, Y.; Shadel, G.S. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007, 5, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Zarse, K.; Schmeisser, S.; Groth, M.; Priebe, S.; Beuster, G.; Kuhlow, D.; Guthke, R.; Platzer, M.; Kahn, C.R.; Ristow, M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial l-proline catabolism to induce a transient ROS signal. Cell Metab. 2012, 15, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Hamadeh, M.J.; Kaczor, J.J.; Raha, S.; Debeer, J.; Tarnopolsky, M.A. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 2010, 5, e10778. [Google Scholar] [CrossRef] [PubMed]
- Menshikova, E.V.; Ritov, V.B.; Fairfull, L.; Ferrell, R.E.; Kelley, D.E.; Goodpaster, B.H. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J. Gerontol. A 2006, 61, 534–540. [Google Scholar] [CrossRef]
- Lee, S.J.; Hwang, A.B.; Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.; Yang, W.; Hekimi, S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 2014, 157, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Rauthan, M.; Ranji, P.; Aguilera Pradenas, N.; Pitot, C.; Pilon, M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.; Talloczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, O.; Leduc-Gaudet, J.P.; Hussain, S.; Gouspillou, G. Parkin Overexpression Attenuates Aging-related Muscle Atrophy in Mice. FASEB J. 2017, 31, 713.11. [Google Scholar]
- Rodriguez-Navarro, J.A.; Casarejos, M.J.; Menendez, J.; Solano, R.M.; Rodal, I.; Gomez, A.; Yebenes, J.G.; Mena, M.A. Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J. Neurochem. 2007, 103, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Voloboueva, L.A.; Lee, S.W.; Emery, J.F.; Palmer, T.D.; Giffard, R.G. Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J. Neurosci. 2010, 30, 12242–12251. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Guimera, A.M.; Hodgson, D.; McDonald, N.; Mooney, K.M.; Morgan, A.E.; Proctor, C.J. Modelling the molecular mechanisms of aging. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.S.; French, D.P.; Jackson, D.; Nazroo, J.; Pendleton, N.; Degens, H. Physical activity in older age: Perspectives for healthy ageing and frailty. Biogerontology 2016, 17, 567–580. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theurey, P.; Pizzo, P. The Aging Mitochondria. Genes 2018, 9, 22. https://doi.org/10.3390/genes9010022
Theurey P, Pizzo P. The Aging Mitochondria. Genes. 2018; 9(1):22. https://doi.org/10.3390/genes9010022
Chicago/Turabian StyleTheurey, Pierre, and Paola Pizzo. 2018. "The Aging Mitochondria" Genes 9, no. 1: 22. https://doi.org/10.3390/genes9010022
APA StyleTheurey, P., & Pizzo, P. (2018). The Aging Mitochondria. Genes, 9(1), 22. https://doi.org/10.3390/genes9010022