
MYC Deregulation in Primary Human Cancers

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
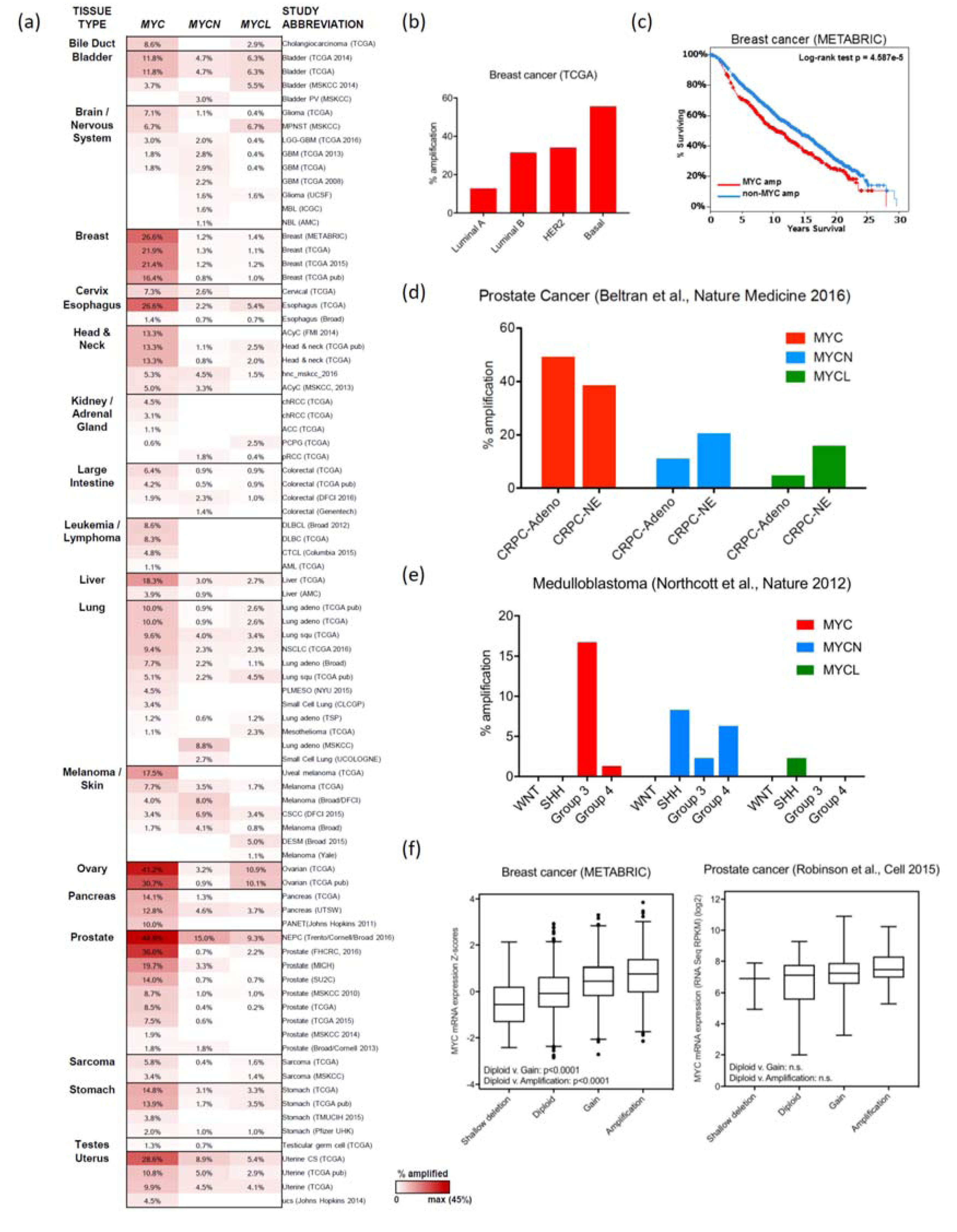
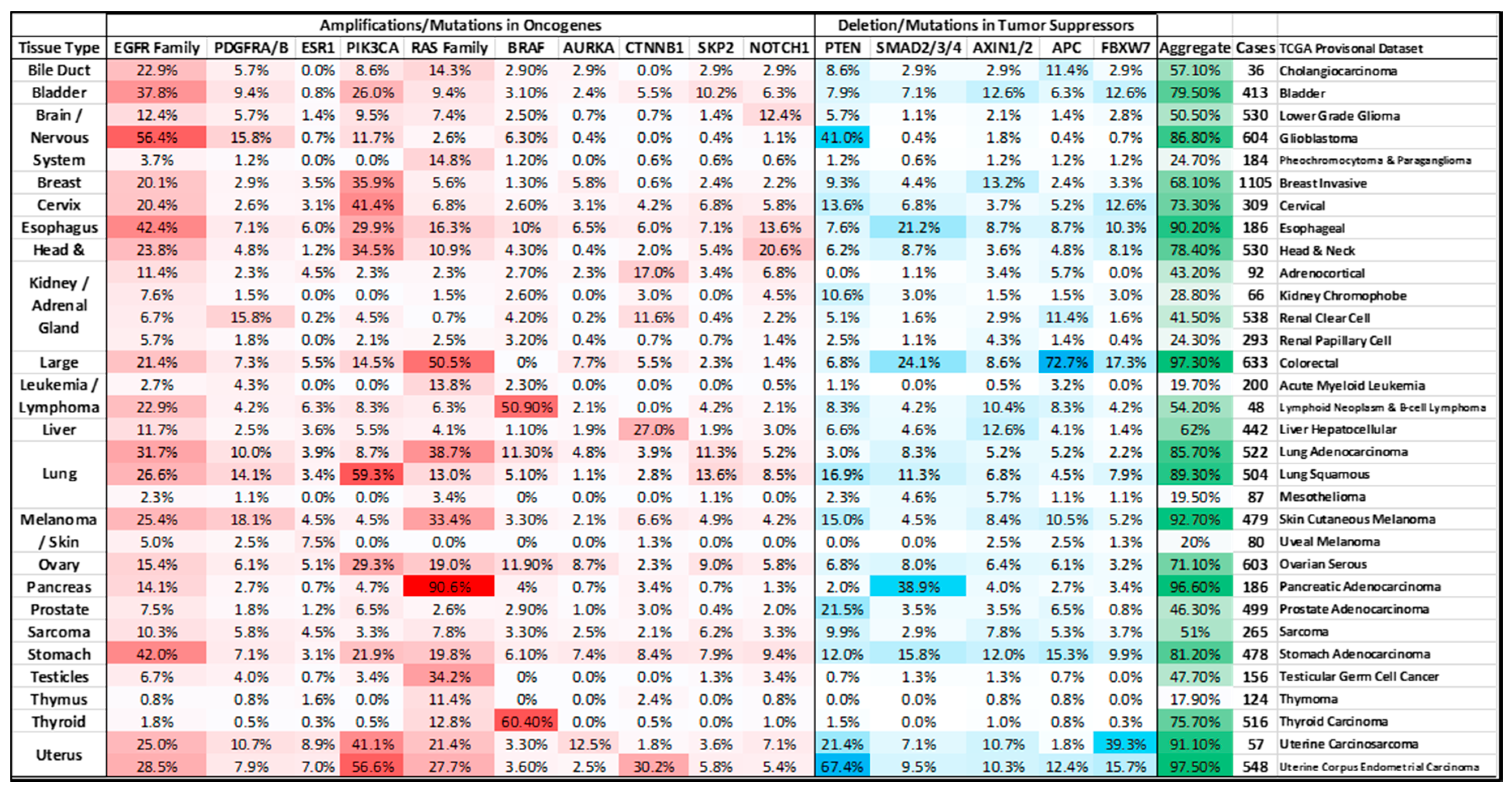
2. MYC Amplification
2.1. Breast Cancer
2.2. Ovarian and Endometrial Cancers
2.3. Colorectal Cancer
2.4. Prostate Cancer
2.5. Lung Cancer
2.6. Pancreatic Cancer
2.7. Renal Clear Cell Carcinoma and Adrenal Cell Carcinoma
2.8. MYC CNAs in Pediatric Cancers
2.8.1. Medulloblastoma
2.8.2. Neuroblastoma
2.9. MAX Alterations
2.10. Considerations for MYC Amplification
3. Enhancer Activity
3.1. Translocations
3.2. Activated Enhancer Activity
3.2.1. Single Nucleotide Polymorphisms and MYC Transcription
3.2.2. Activation of Enhancer Sequences
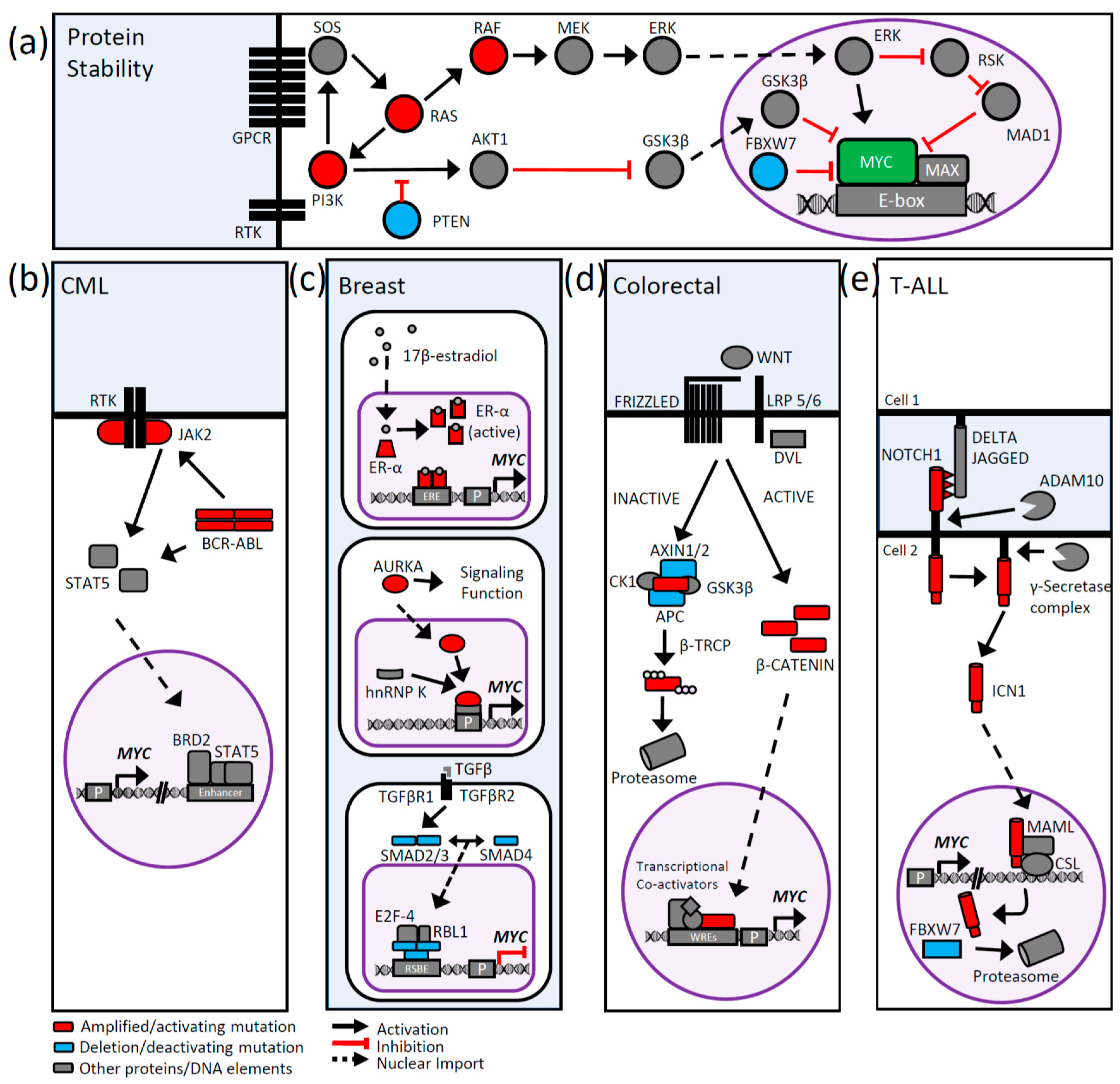
4. MYC Protein Stability
4.1. MYC Protein Turnover
4.2. MYC Mutations and Protein Stability
5. Signaling Pathways and MYC Deregulation
5.1. PI3K/AKT Signaling
5.2. MAPK Signaling
5.3. BCR-ABL1 Signaling in Chronic Myeloid Leukemia
5.4. Breast Cancer Signaling and MYC
5.5. WNT Signaling and MYC
5.6. NOTCH Signaling and MYC
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptional activation by the human c-MYC oncoprotein in yeast requires interaction with MAX. Nature 1992, 359, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-MYC protein requires dimerization with MAX. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Penn, L.J.; Brooks, M.W.; Laufer, E.M.; Land, H. Negative autoregulation of c-MYC transcription. EMBO J. 1990, 9, 1113–1121. [Google Scholar] [PubMed]
- Mao, D.Y.; Watson, J.D.; Yan, P.S.; Barsyte-Lovejoy, D.; Khosravi, F.; Wong, W.W.; Farnham, P.J.; Huang, T.H.; Penn, L.Z. Analysis of MYC bound loci identified by CpG island arrays shows that MAX is essential for MYC-dependent repression. Curr. Biol. 2003, 13, 882–886. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Obaya, A.J.; Adachi, S.; Sedivy, J.M. Phenotypes of c-MYC-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997, 8, 1039–1048. [Google Scholar] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c-MYC gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Eberhardy, S.R.; Farnham, P.J. C-MYC mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem. 2001, 276, 48562–48571. [Google Scholar] [CrossRef] [PubMed]
- Eberhardy, S.R.; Farnham, P.J. MYC recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J. Biol. Chem. 2002, 277, 40156–40162. [Google Scholar] [CrossRef] [PubMed]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. C-MYC regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Cole, M.D. MYC regulation of mRNA cap methylation. Genes Cancer 2010, 1, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Cole, M.D. The MYC transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol. Cell. Biol. 2007, 27, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-MYC. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, M.; Bakardjiev, A.; Klein, G.; Luscher, B. Phosphorylation sites mapping in the N-terminal domain of c-MYC modulate its transforming potential. Oncogene 1993, 8, 3199–3209. [Google Scholar] [PubMed]
- Lutterbach, B.; Hann, S.R. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-MYC protein is regulated by mitogens and in mitosis. Mol. Cell. Biol. 1994, 14, 5510–5522. [Google Scholar] [CrossRef] [PubMed]
- Hasmall, S.C.; Pyrah, I.T.; Soames, A.R.; Roberts, R.A. Expression of the immediate-early genes, c-FOS, c-JUN, and c-MYC: A comparison in rats of nongenotoxic hepatocarcinogens with noncarcinogenic liver mitogens. Fundam. Appl. Toxicol. 1997, 40, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Groudine, M. Amplification of endogenous MYC-related DNA sequences in a human myeloid leukaemia cell line. Nature 1982, 298, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to MYC cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Nau, M.M.; Brooks, B.J.; Battey, J.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.; Hollis, G.F.; Minna, J.D. L-MYC, a new MYC-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and expression of the c-MYC oncogene in human lung cancer cell lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Nau, M.M.; Brooks, B.J., Jr.; Carney, D.N.; Gazdar, A.F.; Battey, J.F.; Sausville, E.A.; Minna, J.D. Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Wardell, C.P.; Furuta, M.; Taniguchi, H.; Fujimoto, A. Cancer whole-genome sequencing: Present and future. Oncogene 2015, 34, 5943–5950. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sanchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Murthy, M.S.; Pande, S.V. Characterization of a solubilized malonyl-CoA-sensitive carnitine palmitoyltransferase from the mitochondrial outer membrane as a protein distinct from the malonyl-CoA-insensitive carnitine palmitoyltransferase of the inner membrane. Biochem. J. 1990, 268, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Medic. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-MYC is mediated by the F-box protein FBW7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The FBW7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-MYC protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [PubMed]
- Calonge, M.J.; Massague, J. SMAD4/DPC4 silencing and hyperactive ras jointly disrupt transforming growth factor-β antiproliferative responses in colon cancer cells. J. Biol. Chem. 1999, 274, 33637–33643. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGFβ influences Myc, Miz-1 and smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Nagl, N.G., Jr.; Zweitzig, D.R.; Thimmapaya, B.; Beck, G.R., Jr.; Moran, E. The c-MYC gene is a direct target of mammalian SWI/SNF-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 2006, 66, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [PubMed]
- Berruti, A.; Pia, A.; Terzolo, M. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 365, 766; author reply 767–768. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Berns, A. Cell of origin of lung cancer. Mol. Oncol. 2010, 4, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Jager, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stutz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Erkek, S.; Tong, Y.; Yin, L.; Federation, A.J.; Zapatka, M.; Haldipur, P.; Kawauchi, D.; Risch, T.; Warnatz, H.J.; et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016, 530, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, D.; Robinson, G.; Uziel, T.; Gibson, P.; Rehg, J.; Gao, C.; Finkelstein, D.; Qu, C.; Pounds, S.; Ellison, D.W.; et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 2012, 21, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and p53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-MYC in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Schleiermacher, G.; Michels, E.; Mosseri, V.; Ribeiro, A.; Lequin, D.; Vermeulen, J.; Couturier, J.; Peuchmaur, M.; Valent, A.; et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. 2009, 27, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. MAX: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with MYC. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Ribon, V.; Leff, T.; Saltiel, A.R. C-MYC does not require MAX for transcriptional activity in PC-12 cells. Mol. Cell. Neurosci. 1994, 5, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hopewell, R.; Ziff, E.B. The nerve growth factor-responsive PC12 cell line does not express the MYC dimerization partner MAX. Mol. Cell. Biol. 1995, 15, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Comino-Mendez, I.; Gracia-Aznarez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.J.; Leton, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Romero, O.A.; Torres-Diz, M.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Saez, C.; Iwakawa, R.; Villanueva, A.; Montuenga, L.M.; et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov. 2014, 4, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, A.; Idbaih, A.; Dehais, C.; Elarouci, N.; Carpentier, C.; Letouze, E.; Colin, C.; Mokhtari, K.; Jouvet, A.; Uro-Coste, E.; et al. Integrated multi-omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co-deleted gliomas. Nat. Commun. 2016, 7, 11263. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Escot, C.; Theillet, C.; Lidereau, R.; Spyratos, F.; Champeme, M.H.; Gest, J.; Callahan, R. Genetic alteration of the c-MYC protooncogene (MYC) in human primary breast carcinomas. Proc. Natl. Acad. Sci. USA 1986, 83, 4834–4838. [Google Scholar] [CrossRef] [PubMed]
- Mariani-Costantini, R.; Escot, C.; Theillet, C.; Gentile, A.; Merlo, G.; Lidereau, R.; Callahan, R. In situ c-MYC expression and genomic status of the c-MYC locus in infiltrating ductal carcinomas of the breast. Cancer Res. 1988, 48, 199–205. [Google Scholar] [PubMed]
- Slavc, I.; Ellenbogen, R.; Jung, W.H.; Vawter, G.F.; Kretschmar, C.; Grier, H.; Korf, B.R. MYC gene amplification and expression in primary human neuroblastoma. Cancer Res. 1990, 50, 1459–1463. [Google Scholar] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. C-MYC is an important direct target of NOTCH1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple RAS-dependent phosphorylation pathways regulate MYC protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Ulz, P.; Heitzer, E.; Speicher, M.R. Co-occurrence of MYC amplification and TP53 mutations in human cancer. Nat. Genet. 2016, 48, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; De Miglio, M.R.; Muroni, M.R.; Simile, M.M.; Daino, L.; Seddaiu, M.A.; Nufris, A.; Gaspa, L.; Deiana, L.; Feo, F. C-MYC amplification in pre-malignant and malignant lesions induced in rat liver by the resistant hepatocyte model. Int. J. Cancer 1996, 68, 136–142. [Google Scholar] [CrossRef]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern MYC’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-MYC protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.H.; Iyer, V.R. Global identification of MYC target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS ONE 2008, 3, e1798. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Lewis, B.C.; Dolde, C.; Li, Q.; Wu, C.S.; Chun, Y.S.; Dang, C.V. MYC target genes in neoplastic transformation. Curr. Top. Microbiol. Immunol. 1997, 224, 181–190. [Google Scholar] [PubMed]
- Dang, C.V. C-MYC target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-MYC onc gene is located on the region of chromosome 8 that is translocated in burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Kaiser-McCaw, B.; Epstein, A.L.; Kaplan, H.S.; Hecht, F. Chromosome 14 translocation in african and north american burkitt’s lymphoma. Int. J. Cancer 1977, 19, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-MYC gene into the immunoglobulin heavy chain locus in human burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed]
- Marcu, K.B.; Harris, L.J.; Stanton, L.W.; Erikson, J.; Watt, R.; Croce, C.M. Transcriptionally active c-MYC oncogene is contained within NIARD, a DNA sequence associated with chromosome translocations in B-cell neoplasia. Proc. Natl. Acad. Sci. USA 1983, 80, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.; Munshi, N.C.; Anderson, K.C. Focus on multiple myeloma. Cancer Cell 2004, 6, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Fonseca, R. Prognostic and therapeutic significance of myeloma genetics and gene expression profiling. J. Clin. Oncol. 2005, 23, 6339–6344. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J. Multiple myeloma. Clin. J. Am. Soc. Nephrol. 2006, 1, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC translocations in multiple myeloma cell lines. J. Natl. Cancer Inst. Monogr. 2008, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Jara-Acevedo, M.; Corchete, L.A.; Castillo, D.; Ordonez, G.R.; Sarasquete, M.E.; Puig, N.; Martinez-Lopez, J.; Prieto-Conde, M.I.; Garcia-Alvarez, M.; et al. A next-generation sequencing strategy for evaluating the most common genetic abnormalities in multiple myeloma. J. Mol. Diagn. 2017, 19, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Grisanzio, C.; Freedman, M.L. Chromosome 8q24-associated cancers and MYC. Genes Cancer 2010, 1, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Eeles, R.A. Genome-wide association studies in cancer. Hum. Mol. Genet. 2008, 17, R109–R115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.; Zotenko, E.; Locke, W.J.; Korbie, D.; Millar, E.K.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 2015, 6, 7758. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Vidal, E.; Ferreira, H.J.; Vizoso, M.; Sayols, S.; Gomez, A.; Moran, S.; Boque-Sastre, R.; Guil, S.; Martinez-Cardus, A.; et al. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Corradin, O.; Scacheri, P.C. Enhancer variants: Evaluating functions in common disease. Genome Med. 2014, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.B.; Brown, S.J.; Cole, M.D. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell. Biol. 2010, 30, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Patterson, N.; Freedman, M.L.; Myers, S.R.; Pike, M.C.; Waliszewska, A.; Neubauer, J.; Tandon, A.; Schirmer, C.; McDonald, G.J.; et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat. Genet. 2007, 39, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010, 20, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Oktay, Y.; Ulgen, E.; Can, O.; Akyerli, C.B.; Yuksel, S.; Erdemgil, Y.; Durasi, I.M.; Henegariu, O.I.; Nanni, E.P.; Selevsek, N.; et al. IDH-mutant glioma specific association of rs55705857 located at 8q24.21 involves MYC deregulation. Sci. Rep. 2016, 6, 27569. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Webb, E.; Carvajal-Carmona, L.; Broderick, P.; Howarth, K.; Pittman, A.M.; Spain, S.; Lubbe, S.; Walther, A.; Sullivan, K.; et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet. 2008, 40, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Le Marchand, L.; Yamamato, J.; Stram, D.O.; Sheng, X.; Kolonel, L.N.; Wu, A.H.; Reich, D.; Henderson, B.E. A common genetic risk factor for colorectal and prostate cancer. Nat. Genet. 2007, 39, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009, 41, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced WNT signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated MYC regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Pinz, S.; Unser, S.; Rascle, A. Signal transducer and activator of transcription STAT5 is recruited to c-MYC super-enhancer. BMC Mol. Biol. 2016, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Radtke, I.; Mullighan, C.G.; Ishii, M.; Su, X.; Cheng, J.; Ma, J.; Ganti, R.; Cai, Z.; Goorha, S.; Pounds, S.B.; et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 12944–12949. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.W.; Radtke, I.; Bullinger, L.; Goorha, S.; Cheng, J.; Edelmann, J.; Gohlke, J.; Su, X.; Paschka, P.; Pounds, S.; et al. High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 2012, 119, e67–e75. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Rennoll, S.; Yochum, G. Regulation of MYC gene expression by aberrant WNT/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Yochum, G.S. Multiple WNT/β-catenin responsive enhancers align with the MYC promoter through long-range chromatin loops. PLoS ONE 2011, 6, e18966. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navas, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.J.; Drier, Y.; Whitton, H.; Cotton, M.J.; Kaur, J.; Issner, R.; Gillespie, S.; Epstein, C.B.; Nardi, V.; Sohani, A.R.; et al. Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov. 2015, 5, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Affer, M.; Chesi, M.; Chen, W.D.; Keats, J.J.; Demchenko, Y.N.; Tamizhmani, K.; Garbitt, V.M.; Riggs, D.L.; Brents, L.A.; Roschke, A.V.; et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. Bet bromodomain inhibition as a therapeutic strategy to target c-MYC. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-MYC proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-MYC degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Deng, Q.; Jiang, C.; Wang, X.; Niu, T.; Li, H.; Chen, T.; Jin, J.; Pan, W.; Cai, X.; et al. USP37 directly deubiquitinates and stabilizes c-MYC in lung cancer. Oncogene 2015, 34, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hong, A.; Park, H.I.; Shin, W.H.; Yoo, L.; Jeon, S.J.; Chung, K.C. Deubiquitinating enzyme USP22 positively regulates c-MYC stability and tumorigenic activity in mammalian and breast cancer cells. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Diefenbacher, M.E.; Popov, N.; Blake, S.M.; Schulein-Volk, C.; Nye, E.; Spencer-Dene, B.; Jaenicke, L.A.; Eilers, M.; Behrens, A. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J. Clin. Investig. 2014, 124, 3407–3418. [Google Scholar] [CrossRef] [PubMed]
- Akhoondi, S.; Sun, D.; von der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dafou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; He, X.; Yin, L.; Komada, M.; Sears, R.C.; Dai, M.S. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-MYC. Proc. Natl. Acad. Sci. USA 2015, 112, 3734–3739. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Grim, J.E.; Eisenman, R.N.; Clurman, B.E. A nucleolar isoform of the FBW7 ubiquitin ligase regulates c-MYC and cell size. Curr. Biol. 2004, 14, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. SKP2 regulates MYC protein stability and activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar] [CrossRef]
- Kimura, Y.; Nagao, A.; Fujioka, Y.; Satou, A.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. MM-1 facilitates degradation of c-MYC by recruiting proteasome and a novel ubiquitin E3 ligase. Int. J. Oncol. 2007, 31, 829–836. [Google Scholar] [CrossRef] [PubMed]
- von der Lehr, N.; Johansson, S.; Larsson, L.G. Implication of the ubiquitin/proteasome system in MYC-regulated transcription. Cell Cycle 2003, 2, 403–407. [Google Scholar] [PubMed]
- Gstaiger, M.; Jordan, R.; Lim, M.; Catzavelos, C.; Mestan, J.; Slingerland, J.; Krek, W. SKP2 is oncogenic and overexpressed in human cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 5043–5048. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt’s lymphoma-associated c-MYC mutations converge on a dramatically altered target gene response and implicate NOL5A/NOP56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.; Huppi, K.; Spangler, G.; Siwarski, D.; Iyer, R.; Magrath, I. Point mutations in the c-MYC transactivation domain are common in burkitt’s lymphoma and mouse plasmacytomas. Nat. Genet. 1993, 5, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Bahram, F.; von der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-MYC hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110. [Google Scholar] [PubMed]
- Symonds, G.; Hartshorn, A.; Kennewell, A.; O’Mara, M.A.; Bruskin, A.; Bishop, J.M. Transformation of murine myelomonocytic cells by MYC: Point mutations in v-MYC contribute synergistically to transforming potential. Oncogene 1989, 4, 285–294. [Google Scholar] [PubMed]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.W.; Claassen, G.F.; Hann, S.R.; Cole, M.D. The c-MYC transactivation domain is a direct modulator of apoptotic versus proliferative signals. Mol. Cell. Biol. 2000, 20, 4309–4319. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.A.; Scuoppo, C.; Dey, S.; Thomas, L.R.; Lorey, S.L.; Lowe, S.W.; Tansey, W.P. A common functional consequence of tumor-derived mutations within c-MYC. Oncogene 2015, 34, 2406–2409. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-MYC binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Stokoe, D.; Stephens, L.R.; Copeland, T.; Gaffney, P.R.; Reese, C.B.; Painter, G.F.; Holmes, A.B.; McCormick, F.; Hawkins, P.T. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 1997, 277, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Pass, I.; Batty, I.H.; Van der Kaay, J.; Stolarov, J.P.; Hemmings, B.A.; Wigler, M.H.; Downes, C.P.; Tonks, N.K. The lipid phosphatase activity of pten is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA 1998, 95, 13513–13518. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. Pi3k-akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Chardin, P.; Camonis, J.H.; Gale, N.W.; van Aelst, L.; Schlessinger, J.; Wigler, M.H.; Bar-Sagi, D. Human SOS1: A guanine nucleotide exchange factor for ras that binds to GRB2. Science 1993, 260, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.K.; Neel, N.F.; Hu, C.; Gautam, P.; Chenard, M.; Long, B.; Aziz, M.; Kassner, M.; Bryant, K.L.; Pierobon, M.; et al. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 2016, 29, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Hawkins, P.; Stephens, L. Activation of phosphoinositide 3-kinase gamma by RAS. Curr. Biol. 2002, 12, 1068–1075. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of RAS. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Kodaki, T.; Woscholski, R.; Hallberg, B.; Rodriguez-Viciana, P.; Downward, J.; Parker, P.J. The activation of phosphatidylinositol 3-kinase by RAS. Curr. Biol. 1994, 4, 798–806. [Google Scholar] [CrossRef]
- Rottmann, S.; Luscher, B. The mad side of the MAX network: Antagonizing the function of MYC and more. Curr. Top. Microbiol. Immunol. 2006, 302, 63–122. [Google Scholar] [PubMed]
- Ayer, D.E.; Kretzner, L.; Eisenman, R.N. Mad: A heterodimeric partner for MAX that antagonizes MYC transcriptional activity. Cell 1993, 72, 211–222. [Google Scholar] [CrossRef]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/AKT and MAPK pathways regulates MYC-mediated transcription by phosphorylating and promoting the degradation of MAD1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of RAS mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.B.; Schaber, M.D.; Allard, W.J.; Sigal, I.S.; Scolnick, E.M. Purification of RAS GTPase activating protein from bovine brain. Proc. Natl. Acad. Sci. USA 1988, 85, 5026–5030. [Google Scholar] [CrossRef] [PubMed]
- Vogel, U.S.; Dixon, R.A.; Schaber, M.D.; Diehl, R.E.; Marshall, M.S.; Scolnick, E.M.; Sigal, I.S.; Gibbs, J.B. Cloning of bovine GAP and its interaction with oncogenic RAS p21. Nature 1988, 335, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Franken, S.M.; Scheidig, A.J.; Krengel, U.; Rensland, H.; Lautwein, A.; Geyer, M.; Scheffzek, K.; Goody, R.S.; Kalbitzer, H.R.; Pai, E.F.; et al. Three-dimensional structures and properties of a transforming and a nontransforming glycine-12 mutant of p21H-RAS. Biochemistry 1993, 32, 8411–8420. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Gilsbach, B.; Ahmadian, M.R.; Wittinghofer, A. Fluoride complexes of oncogenic RAS mutants to study the RAS-RASGAP interaction. Biol. Chem. 2008, 389, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the braf gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, D.; Ruggiero, A.; Amato, M.; Maurizi, P.; Riccardi, R. BRAF and MEK inhibitors in pediatric glioma: New therapeutic strategies, new toxicities. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Bartram, C.R.; Klein, A.D.; Hagemeijer, A.; Agthoven, T.V.; Kessel, A.G.V.; Bootsma, D.; Grosveld, G.; Ferguson-Smith, M.A.; Davies, T.; Stone, M. Translocation of c-ab1 oncogene correlates with the presence of a philadelphia chromosome in chronic myelocytic leukaemia. Nature 1983, 306, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L.; Callahan, W.; Witte, O.N. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992, 70, 901–910. [Google Scholar] [CrossRef]
- Nieborowska-Skórska, M.; Ratajczak, M.Z.; Calabretta, B.; Skórski, T. The role of c-MYC protooncogene in chronic myelogenous leukemia. Folia Histochem. Cytobiol. 1994, 32, 231–234. [Google Scholar] [PubMed]
- Miyamoto, N.; Sugita, K.; Goi, K.; Inukai, T.; Lijima, K.; Tezuka, T.; Kojika, S.; Nakamura, M.; Kagami, K.; Nakazawa, S. The JAK2 inhibitor AG490 predominantly abrogates the growth of human B-precursor leukemic cells with 11q23 translocation or philadelphia chromosome. Leukemia 2001, 15, 1758–1768. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, K.; Nosaka, T.; Yamada, K.; Onishi, M.; Oka, Y.; Miyajima, A.; Kitamura, T. Constitutive activation of STAT5 by a point mutation in the SH2 domain. J. Biol. Chem. 2000, 275, 24407–24413. [Google Scholar] [CrossRef] [PubMed]
- Oliver, H.; Wolfgang, W.; Eva, E.; Ines, K.; Florian, G.; Kay-Uwe, W.; Giulio, S.-F.; Veronika, S. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 2012, 8, 285–293. [Google Scholar]
- Lord, J.D.; McIntosh, B.C.; Greenberg, P.D.; Nelson, B.H. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-MYC, Bcl-2, and Bcl-x genes through the trans-activation domain of STAT5. J. Immunol. 2000, 164, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, T.; Kawashima, T.; Misawa, K.; Ikuta, K.; Mui, A.L.; Kitamura, T. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 1999, 18, 4754–4765. [Google Scholar] [CrossRef] [PubMed]
- Beth, B.; Manjiri, S.; Jeffrey, G.; Terrill, M.; Annalisa, D.A.; Emma, L.; Anne, R. In vivo identification of novel STAT5 target genes. Nucleic Acids Res. 2008, 36, 3802–3818. [Google Scholar]
- Pinz, S.; Unser, S.; Buob, D.; Fischer, P.; Jobst, B.; Rascle, A. Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (bet) protein function. Nucleic Acids Res. 2015, 43, 3524–3545. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R. Genetic complexity of myeloproliferative neoplasms. Leukemia 2008, 22, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Couédic, J.-P.L.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Dogara, L.G.; Babadoko, A.A.; Awwalu, S.; Mamman, A.I. Coexistence of JAK2 and BCR-ABL mutation in patient with myeloproliferative neoplasm. Niger. Med. J. 2015, 56, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Pahore, Z.-A.-A.; Shamsi, T.S.; Taj, M.; Farzana, T.; Ansari, S.H.; Nadeem, M.; Ahmad, M.; Naz, A. JAK2V617F mutation in chronic myeloid leukemia predicts early disease progression. J. Coll. Phys. Surg. Pak. 2011, 21, 472–475. [Google Scholar]
- Dubik, D.; Dembinski, T.C.; Shiu, R.P. Stimulation of c-MYC oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987, 47, 6517–6521. [Google Scholar] [PubMed]
- Dubik, D.; Shiu, R.P. Mechanism of estrogen activation of c-MYC oncogene expression. Oncogene 1992, 7, 1587–1594. [Google Scholar] [PubMed]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.J.; Caldon, C.E.; McNeil, C.M.; Alexander, S.; Musgrove, E.A.; Sutherland, R.L. Cell cycle machinery: Links with genesis and treatment of breast cancer. Adv. Exp. Med. Biol. 2008, 630, 189–205. [Google Scholar] [PubMed]
- Alles, M.C.; Margaret, G.-G.; David, J.N.; Yixin, W.; John, A.F.; Robert, L.S.; Elizabeth, A.M.; Christopher, J.O. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PLoS ONE 2009, 4, e4710. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sergio, C.M.; Loi, S.; Inman, C.K.; Anderson, L.R.; Alles, M.C.; Pinese, M.; Caldon, C.E.; Schütte, J.; Gardiner-Garden, M.; et al. Identification of functional networks of estrogen- and c-MYC-responsive genes and their relationship to response to tamoxifen therapy in breast cancer. PLoS ONE 2008, 3, e2987. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Frengen, E.; Cowling, V.H.; Pendergrass, S.A.; Perou, C.M.; Whitfield, M.L.; Cole, M.D. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS ONE 2009, 4, e6693. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kimura, M.; Matsunaga, K.; Fukada, D.; Mori, H.; Okano, Y. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999, 59, 2041–2044. [Google Scholar] [PubMed]
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear aurka acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.-L.; Gray, J.; Sahin, A.; Brinkley, B.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Frederick, J.P.; Liberati, N.T.; Waddell, D.S.; Shi, Y.; Wang, X.-F. Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol. Cell. Biol. 2004, 24, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Furuhashi, M.; Aoki, H.; Goto, D.; Kuwano, H.; Sugamura, K.; Miyazono, K.; Kato, M. C-MYC is a downstream target of the SMAD pathway. J. Biol. Chem. 2001, 277, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Jeruss, J.S.; Sturgis, C.D.; Rademaker, A.W.; Woodruff, T.K. Down-regulation of activin, activin receptors, and SMADS in high-grade breast cancer. Cancer Res. 2003, 63, 3783–3790. [Google Scholar] [PubMed]
- Sekimoto, G.; Matsuzaki, K.; Yoshida, K.; Mori, S.; Murata, M.; Seki, T.; Matsui, H.; Fujisawa, J.-I.; Okazaki, K. Reversible SMAD-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007, 67, 5090–5096. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kang, Y.; Massagué, J. Defective repression of c-MYC in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc. Natl. Acad. Sci. USA 2001, 98, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, Y.X.; Yu, J.H.; Chen, X.; Luo, X.J.; Yin, Y.R. Research of the relationship between β-catenin and c-myc-mediated Wnt pathway and laterally spreading tumors occurrence. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 252–257. [Google Scholar] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. WNT signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.; Sansom, O.J. WNT/MYC interactions in intestinal cancer: Partners in crime. Exp. Cell Res. 2011, 317, 2725–2731. [Google Scholar] [CrossRef] [PubMed]
- Najdi, R.; Holcombe, R.F.; Waterman, M.L. WNT signaling and colon carcinogenesis: Beyond APC. J. Carcinog. 2011, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, X.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating β-catenin/TCF signalling. Nat. Genet. 2000, 26, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014, 355, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, J.M.; Wang, H.; Kulkarni, S.; Jackson, L.; Lu, J.; Ranganathan, S.; Goetzman, E.S.; Bharathi, S.; Beezhold, K.; Byersdorfer, C.A.; et al. Sequential adaptive changes in a c-MYC-driven model of hepatocellular carcinoma. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Koch, A.; Uhlhaas, S.; Friedl, W.; Propping, P.; Schweinitz, D.V.; Pietsch, T. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr. Blood Cancer 2006, 47, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; Armengol, C.; De Reynies, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of WNT/β-catenin and MYC signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, J.; Edmunds, L.R.; Kulkarni, S.; Dolezal, J.; Tao, J.; Ranganathan, S.; Jackson, L.; Fromherz, M.; Beer-Stolz, D.; et al. Coordinated activities of multiple MYC-dependent and MYC-independent biosynthetic pathways in hepatoblastoma. J. Biol. Chem. 2016, 291, 26241–26251. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yang, Y.; Han, B.; Du, C.; Xu, N.; Huang, H.; Cai, T.; Zhang, A.; Han, Z.G.; Zhou, W.; et al. Transcriptomic characterization of hepatocellular carcinoma with CTNNB1 mutation. PLoS ONE 2014, 9, e95307. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway towards T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Buonamici, S.; Sulis, M.L.; Palomero, T.; Vilimas, T.; Basso, G.; Ferrando, A.; Aifantis, I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007, 204, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Wasylishen, A.R.; Chan-Seng-Yue, M.; Bros, C.; Dingar, D.; Tu, W.B.; Kalkat, M.; Chan, P.K.; Mullen, P.J.; Huang, L.; Meyer, N.; et al. MYC phosphorylation at novel regulatory regions suppresses transforming activity. Cancer Res. 2013, 73, 6504–6515. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Traugh, J.A.; Bishop, J.M. Negative control of the MYC protein by the stress-responsive kinase PAK2. Mol. Cell. Biol. 2004, 24, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, B.J.; Fisher, C.; Vousden, K.; Littlewood, T.; Evan, G.; Woodgett, J.R. Site-specific modulation of c-MYC cotransformation by residues phosphorylated in vivo. Oncogene 1994, 9, 59–70. [Google Scholar] [PubMed]
- Luscher, B.; Kuenzel, E.A.; Krebs, E.G.; Eisenman, R.N. MYC oncoproteins are phosphorylated by casein kinase II. EMBO J 1989, 8, 1111–1119. [Google Scholar] [PubMed]
- Kalkat, M.; Chan, P.K.; Wasylishen, A.R.; Srikumar, T.; Kim, S.S.; Ponzielli, R.; Bazett-Jones, D.P.; Raught, B.; Penn, L.Z. Identification of c-MYC sumoylation by mass spectrometry. PLoS ONE 2014, 9, e115337. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Prieto, R.; Cuijpers, S.A.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C. c-MYC is targeted to the proteasome for degradation in a sumoylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Doni, M.; Amati, B. Sumoylation of MYC-family proteins. PLoS ONE 2014, 9, e91072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Faiola, F.; Martinez, E. Six lysine residues on c-MYC are direct substrates for acetylation by p300. Biochem. Biophys. Res. Commun. 2005, 336, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Farina, A.; Martinez, E. Dual regulation of c-MYC by p300 via acetylation-dependent control of MYC protein turnover and coactivation of MYC-induced transcription. Mol. Cell. Biol. 2005, 25, 10220–10234. [Google Scholar] [CrossRef] [PubMed]
- Phosphositeplus: Myc (human). Available online: http://www.phosphosite.org/proteinAction.action?id=947&showAllSites=true (accessed on 27 February 2017).
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the MYC oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tato, F.; Nasi, S. Omomyc, a potential MYC dominant negative, enhances MYC-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling MYC inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Casey, S.C.; Felsher, D.W. Inactivation of MYC reverses tumorigenesis. J. Intern. Med. 2014, 276, 52–60. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. https://doi.org/10.3390/genes8060151
Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, Penn LZ. MYC Deregulation in Primary Human Cancers. Genes. 2017; 8(6):151. https://doi.org/10.3390/genes8060151
Chicago/Turabian StyleKalkat, Manpreet, Jason De Melo, Katherine Ashley Hickman, Corey Lourenco, Cornelia Redel, Diana Resetca, Aaliya Tamachi, William B. Tu, and Linda Z. Penn. 2017. "MYC Deregulation in Primary Human Cancers" Genes 8, no. 6: 151. https://doi.org/10.3390/genes8060151
APA StyleKalkat, M., De Melo, J., Hickman, K. A., Lourenco, C., Redel, C., Resetca, D., Tamachi, A., Tu, W. B., & Penn, L. Z. (2017). MYC Deregulation in Primary Human Cancers. Genes, 8(6), 151. https://doi.org/10.3390/genes8060151