
DNA Methylation Profiling of Human Prefrontal Cortex Neurons in Heroin Users Shows Significant Difference between Genomic Contexts of Hyper- and Hypomethylation and a Younger Epigenetic Age

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Tissue
2.2. Nuclei Separation by Fluorescence Activated Nuclei Sorting (FANS)
2.3. DNA Methylation Measurement and Analysis
2.4. Estimation of DNA Methylation Age
2.5. Gene Ontology Functional Annotation Analysis
2.6. Gene Expression Comparison between Glu- and GABA Neurons in the Human PFC
3. Results
3.1. mOFC Neurons of Heroin Users Show Younger Epigenetic Age Than Neurons of Non-Addicted Individuals
3.2. Differential DNA Methylation Analysis
3.3. Enrichment of Differentially Methylated Sites in Genomic Features and Regulatory Elements
3.4. Gene Ontology Analysis of Differentially Methylated Genes
3.5. Neuronal Subtype-Specificity of Hyper- or Hypomethylated Genes
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lauren, M.; Rossen, B.B.; Warner, M.; Khan, D.; Chong, Y. Drug Poisoning Mortality: United States, 2002–2014; National Center for Health Statistics, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2015.
- Bryant, W.K.; Galea, S.; Tracy, M.; Markham, P.T.; Tardiff, K.J.; Vlahov, D. Overdose deaths attributed to methadone and heroin in New York City, 1990–1998. Addiction 2004, 99, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Inciardi, J.A.; Surratt, H.L.; Cicero, T.J.; Beard, R.A. Prescription opioid abuse and diversion in an urban community: The results of an ultrarapid assessment. Pain Med. 2009, 10, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Peavy, K.M.; Banta-Green, C.J.; Kingston, S.; Hanrahan, M.; Merrill, J.O.; Coffin, P.O. “Hooked on” prescription-type opiates prior to using heroin: Results from a survey of syringe exchange clients. J. Psychoact. Drugs 2012, 44, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Pollini, R.A.; Banta-Green, C.J.; Cuevas-Mota, J.; Metzner, M.; Teshale, E.; Garfein, R.S. Problematic use of prescription-type opioids prior to heroin use among young heroin injectors. Subst. Abus. Rehabil. 2011, 2, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Verweij, K.J.; Gillespie, N.A.; Heath, A.C.; Lessov-Schlaggar, C.N.; Martin, N.G.; Nelson, E.C.; Slutske, W.S.; Whitfield, J.B.; Lynskey, M.T. The genetics of addiction—A translational perspective. Transl. Psychiatry 2012, 2, e140. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.C.; Agrawal, A.; Heath, A.C.; Bogdan, R.; Sherva, R.; Zhang, B.; Al-Hasani, R.; Bruchas, M.R.; Chou, Y.L.; Demers, C.H.; et al. Evidence of CNIH3 involvement in opioid dependence. Mol. Psychiatry 2015, 21, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Kendler, K.S.; Jacobson, K.C.; Prescott, C.A.; Neale, M.C. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am. J. Psychiatry 2003, 160, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J. Epigenetic mechanisms of drug addiction. Neuropharmacology 2014, 76, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.; Truong, H.T.; Russo, S.J.; Laplant, Q.; Sasaki, T.S.; Whistler, K.N.; et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Kumar, A.; Xiao, G.; Wilkinson, M.; Covington, H.E., 3rd; Maze, I.; Sikder, D.; Robison, A.J.; LaPlant, Q.; Dietz, D.M.; et al. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 2009, 62, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Covington, H.E.; Dietz, D.M., 3rd; Dietz, D.M.; LaPlant, Q.; Renthal, W.; Russo, S.J.; Mechanic, M.; Mouzon, E.; Neve, R.L.; Haggarty, S.J.; et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 2010, 327, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Maze, I.; Dietz, D.M.; Scobie, K.N.; Kennedy, P.J.; Damez-Werno, D.; Neve, R.L.; Zachariou, V.; Shen, L.; Nestler, E.J. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J. Neurosci. 2012, 32, 17454–17464. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Mazei-Robison, M.S.; LaPlant, Q.; Egervari, G.; Braunscheidel, K.M.; Adank, D.N.; Ferguson, D.; Feng, J.; Sun, H.; Scobie, K.N.; et al. Epigenetic basis of opiate suppression of BDNF gene expression in the ventral tegmental area. Nat. Neurosci. 2015, 18, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, T.; Nakashima, K.; Namihira, M.; Ochiai, W.; Uemura, A.; Yanagisawa, M.; Fujita, N.; Nakao, M.; Taga, T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell 2001, 1, 749–758. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Labonte, B.; Suderman, M.; Maussion, G.; Navaro, L.; Yerko, V.; Mahar, I.; Bureau, A.; Mechawar, N.; Szyf, M.; Meaney, M.J. Genome-wide epigenetic regulation by early-life trauma. Arch. Gen. Psychiatry 2012, 69, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Suderman, M.; McGowan, P.O.; Sasaki, A.; Huang, T.C.; Hallett, M.T.; Meaney, M.J.; Turecki, G.; Szyf, M. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Nat. Acad. Sci. USA 2012, 109, 17266–17272. [Google Scholar] [CrossRef] [PubMed]
- LaPlant, Q.; Vialou, V.; Covington, H.E., III; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iniguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Shao, N.; Szulwach, K.E.; Vialou, V.; Huynh, J.; Zhong, C.; Le, T.; Ferguson, D.; Cahill, M.E.; Li, Y.; et al. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat. Neurosci. 2015, 18, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011, 10, 2662–2668. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Pevsner, J.; Sabunciyan, S.; Yolken, R.H.; Webster, M.J.; Dinkins, T.; Callinan, P.A.; Fan, J.B.; Potash, J.B.; Feinberg, A.P. DNA methylation signatures within the human brain. Am. J. Hum. Genet. 2007, 81, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.W.; Ersche, K.D.; Everitt, B.J. Drug addiction and the memory systems of the brain. Ann. N. Y. Acad. Sci. 2008, 1141, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tzschentke, T.M. The medial prefrontal cortex as a part of the brain reward system. Amino Acids 2000, 19, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Van den Oever, M.C.; Spijker, S.; Smit, A.B.; De Vries, T.J. Prefrontal cortex plasticity mechanisms in drug seeking and relapse. Neurosci. Biobehav. Rev. 2010, 35, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.Z.; Volkow, N.D. Drug addiction and its underlying neurobiological basis: Neuroimaging evidence for the involvement of the frontal cortex. Am. J. Psychiatry 2002, 159, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Botelho, M.F.; Relvas, J.S.; Abrantes, M.; Cunha, M.J.; Marques, T.R.; Rovira, E.; Fontes Ribeiro, C.A.; Macedo, T. Brain blood flow SPET imaging in heroin abusers. Ann. N. Y. Acad. Sci. 2006, 1074, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Langleben, D.D.; Ruparel, K.; Elman, I.; Busch-Winokur, S.; Pratiwadi, R.; Loughead, J.; O'Brien, C.P.; Childress, A.R. Acute effect of methadone maintenance dose on brain FMRI response to heroin-related cues. Am. J. Psychiatry 2008, 165, 390–394. [Google Scholar] [CrossRef] [PubMed]
- LaLumiere, R.T.; Kalivas, P.W. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 2008, 28, 3170–3177. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; Kalivas, P.W.; Quirk, G.J. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn. Mem. 2009, 16, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.; LaLumiere, R.T.; Kalivas, P.W. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J. Neurosci. 2008, 28, 6046–6053. [Google Scholar] [CrossRef] [PubMed]
- Gansler, D.A.; Lee, A.K.; Emerton, B.C.; D′Amato, C.; Bhadelia, R.; Jerram, M.; Fulwiler, C. Prefrontal regional correlates of self-control in male psychiatric patients: Impulsivity facets and aggression. Psychiatry Res. 2011, 191, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Zald, D.H.; Andreotti, C. Neuropsychological assessment of the orbital and ventromedial prefrontal cortex. Neuropsychologia 2010, 48, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Manzardo, A.M.; Henkhaus, R.S.; Butler, M.G. Global DNA promoter methylation in frontal cortex of alcoholics and controls. Gene 2012, 498, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, H.; Zhao, H.; Gelernter, J.; Zhang, H. DNA co-methylation modules in postmortem prefrontal cortex tissues of European Australians with alcohol use disorders. Sci. Rep. 2016, 6, 19430. [Google Scholar] [CrossRef] [PubMed]
- Guintivano, J.; Aryee, M.J.; Kaminsky, Z.A. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 2013, 8, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Roussos, P.; Timashpolsky, A.; Barbu, M.; Rudchenko, S.; Bibikova, M.; Klotzle, B.; Byne, W.; Lyddon, R.; Di Narzo, A.F. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014, 42, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, L.; Lenart, A.; Tan, Q.; Vaupel, J.W.; Aviv, A.; McGue, M.; Christensen, K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 2016, 15, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Perna, L.; Zhang, Y.; Mons, U.; Holleczek, B.; Saum, K.U.; Brenner, H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin. Epigenet. 2016, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Pirazzini, C.; Bacalini, M.G.; Gentilini, D.; Di Blasio, A.M.; Delledonne, M.; Mari, D.; Arosio, B.; Monti, D.; Passarino, G.; et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging 2015, 7, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [PubMed]
- Breitling, L.P.; Saum, K.U.; Perna, L.; Schottker, B.; Holleczek, B.; Brenner, H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin. Epigenet. 2016, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Hosgood, H.D.; Chen, B.; Absher, D.; Assimes, T.; Horvath, S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging 2015, 7, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Mah, V.; Lu, A.T.; Woo, J.S.; Choi, O.W.; Jasinska, A.J.; Riancho, J.A.; Tung, S.; Coles, N.S.; Braun, J.; et al. The cerebellum ages slowly according to the epigenetic clock. Aging 2015, 7, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.F.; Liu, J.S.; Peters, B.A.; Ritz, B.R.; Wu, T.; Ophoff, R.A.; Horvath, S. Epigenetic age analysis of children who seem to evade aging. Aging 2015, 7, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Levine, A.J. HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging 2016, 8, 1485–1512. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schonfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Nat. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed]
- Zannas, A.S.; Arloth, J.; Carrillo-Roa, T.; Iurato, S.; Roh, S.; Ressler, K.J.; Nemeroff, C.B.; Smith, A.K.; Bradley, B.; Heim, C.; et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: Relevance of glucocorticoid signaling. Genome Biol. 2015, 16, 266. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Ritz, B.R. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging 2015, 7, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Drakenberg, K.; Nikoshkov, A.; Horvath, M.C.; Fagergren, P.; Gharibyan, A.; Saarelainen, K.; Rahman, S.; Nylander, I.; Bakalkin, G.; Rajs, J.; et al. Mu opioid receptor A118G polymorphism in association with striatal opioid neuropeptide gene expression in heroin abusers. Proc. Nat. Acad. Sci. USA 2006, 103, 7883–7888. [Google Scholar] [CrossRef] [PubMed]
- Nikoshkov, A.; Drakenberg, K.; Wang, X.; Horvath, M.C.; Keller, E.; Hurd, Y.L. Opioid neuropeptide genotypes in relation to heroin abuse: Dopamine tone contributes to reversed mesolimbic proenkephalin expression. Proc. Nat. Acad. Sci. USA 2008, 105, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.A.; Michaelides, M.; Zarnegar, P.; Ren, Y.; Fagergren, P.; Thanos, P.K.; Wang, G.J.; Bannon, M.; Neumaier, J.F.; Keller, E.; et al. Impaired periamygdaloid-cortex prodynorphin is characteristic of opiate addiction and depression. J. Clin. Investig. 2013, 123, 5334–5341. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lu, Z.; Xu, M.; Pan, L.; Deng, Y.; Xie, X.; Liu, H.; Ding, S.; Hurd, Y.L.; Pasternak, G.W.; et al. A heroin addiction severity-associated intronic single nucleotide polymorphism modulates alternative pre-mRNA splicing of the mu opioid receptor gene OPRM1 via hnRNPH interactions. J. Neurosci. 2014, 34, 11048–11066. [Google Scholar] [CrossRef] [PubMed]
- Egervari, G.; Jutras-Aswad, D.; Landry, J.; Miller, M.L.; Anderson, S.A.; Michaelides, M.; Jacobs, M.M.; Peter, C.; Yiannoulos, G.; Liu, X.; et al. A Functional 3′UTR Polymorphism (rs2235749) of Prodynorphin Alters microRNA-365 Binding in Ventral Striatonigral Neurons to Influence Novelty Seeking and Positive Reward Traits. Neuropsychopharmacology 2016, 41, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Okvist, A.; Fagergren, P.; Whittard, J.; Garcia-Osta, A.; Drakenberg, K.; Horvath, M.C.; Schmidt, C.J.; Keller, E.; Bannon, M.J.; Hurd, Y.L. Dysregulated postsynaptic density and endocytic zone in the amygdala of human heroin and cocaine abusers. Biol. Psychiatry 2011, 69, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.M.; Okvist, A.; Horvath, M.; Keller, E.; Bannon, M.J.; Morgello, S.; Hurd, Y.L. Dopamine receptor D1 and postsynaptic density gene variants associate with opiate abuse and striatal expression levels. Mol. Psychiatry 2013, 18, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Sillivan, S.E.; Whittard, J.D.; Jacobs, M.M.; Ren, Y.; Mazloom, A.R.; Caputi, F.F.; Horvath, M.; Keller, E.; Ma'ayan, A.; Pan, Y.X.; et al. ELK1 transcription factor linked to dysregulated striatal mu opioid receptor signaling network and OPRM1 polymorphism in human heroin abusers. Biol. Psychiatry 2013, 74, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Wang, M.; Roussos, P.; Rudchenko, S.; Barbu, M.; Bibikova, M.; Klotzle, B.; Dwork, A.J.; Zhang, B.; Hurd, Y.L.; et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 2016, 44, 2593–2612. [Google Scholar] [CrossRef] [PubMed]
- Cheung, I.; Shulha, H.P.; Jiang, Y.; Matevossian, A.; Wang, J.; Weng, Z.; Akbarian, S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Nat. Acad. Sci. USA 2010, 107, 8824–8829. [Google Scholar] [CrossRef] [PubMed]
- Shulha, H.P.; Crisci, J.L.; Reshetov, D.; Tushir, J.S.; Cheung, I.; Bharadwaj, R.; Chou, H.J.; Houston, I.B.; Peter, C.J.; Mitchell, A.C.; et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 2012, 10, e1001427. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Bundo, M.; Ueda, J.; Oldham, M.C.; Ukai, W.; Hashimoto, E.; Saito, T.; Geschwind, D.H.; Kato, T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011, 21, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Matevossian, A.; Huang, H.S.; Straubhaar, J.; Akbarian, S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 2016, 19, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Le, J.; Barnes, B.; Saedinia-Melnyk, S.; Zhou, L.; Shen, R.; Gunderson, K.L. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics 2009, 1, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Roadmap, E.C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Rudy, B.; Fishell, G.; Lee, S.; Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2011, 71, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.W.; Matsubara, T.; Li, S.; Sengupta, E.J.; Georgiev, D.; Minabe, Y.; Sampson, A.; Hashimoto, T.; Lewis, D.A. Deficits in transcriptional regulators of cortical parvalbumin neurons in schizophrenia. Am. J. Psychiatry 2012, 169, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Hare, B.; Duman, R.S. Prefrontal Cortex GABAergic Deficits and Circuit Dysfunction in the Pathophysiology and Treatment of Chronic Stress and Depression. Curr. Opin. Behav. Sci. 2017, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cobos, I.; Calcagnotto, M.E.; Vilaythong, A.J.; Thwin, M.T.; Noebels, J.L.; Baraban, S.C.; Rubenstein, J.L. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 2005, 8, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Graziane, N.M.; Sun, S.; Wright, W.J.; Jang, D.; Liu, Z.; Huang, Y.H.; Nestler, E.J.; Wang, Y.T.; Schluter, O.M.; Dong, Y. Opposing mechanisms mediate morphine- and cocaine-induced generation of silent synapses. Nat. Neurosci. 2016, 19, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Xuei, X.; Flury-Wetherill, L.; Almasy, L.; Bierut, L.; Tischfield, J.; Schuckit, M.; Nurnberger, J.I., Jr.; Foroud, T.; Edenberg, H.J. Association analysis of genes encoding the nociceptin receptor (OPRL1) and its endogenous ligand (PNOC) with alcohol or illicit drug dependence. Addict. Biol. 2008, 13, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Guzowski, J.F.; Lyford, G.L.; Stevenson, G.D.; Houston, F.P.; McGaugh, J.L.; Worley, P.F.; Barnes, C.A. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci. 2000, 20, 3993–4001. [Google Scholar] [PubMed]
- Lv, X.F.; Sun, L.L.; Cui, C.L.; Han, J.S. NAc Shell Arc/Arg3.1 Protein Mediates Reconsolidation of Morphine CPP by Increased GluR1 Cell Surface Expression: Activation of ERK-Coupled CREB is Required. Int. J. Neuropsychopharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kirk, L.M.; Ti, S.W.; Bishop, H.I.; Orozco-Llamas, M.; Pham, M.; Trimmer, J.S.; Diaz, E. Distribution of the SynDIG4/proline-rich transmembrane protein 1 in rat brain. J. Comp. Neurol. 2016, 524, 2266–2280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gao, X.; Li, C.; Feliciano, C.; Wang, D.; Zhou, D.; Mei, Y.; Monteiro, P.; Anand, M.; Itohara, S.; et al. Impaired Dendritic Development and Memory in Sorbs2 Knock-Out Mice. J. Neurosci. 2016, 36, 2247–2260. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kim, Y.; Han, J.K.; Park, H.; Lee, U.; Na, M.; Jeong, S.; Chung, C.; Cestra, G.; Chang, S. nArgBP2 regulates excitatory synapse formation by controlling dendritic spine morphology. Proc. Nat. Acad. Sci. USA 2016, 113, 6749–6754. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Nestler, E.J. The neural rejuvenation hypothesis of cocaine addiction. Trends Pharmacol. Sci. 2014, 35, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.; Wilcox, H.C.; Eaton, W.W.; Van Eck, K.; Kilaru, V.; Jovanovic, T.; Klengel, T.; Bradley, B.; Binder, E.B.; Ressler, K.J.; et al. Epigenetic and genetic variation at SKA2 predict suicidal behavior and post-traumatic stress disorder. Transl. Psychiatry 2015, 5, e627. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.M.; Crawford, B.; Dempster, E.L.; Hannon, E.; Burrage, J.; Turecki, G.; Kaminsky, Z.; Mill, J. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl. Psychiatry 2017, 7, e989. [Google Scholar] [CrossRef] [PubMed]
- Labonte, B.; Suderman, M.; Maussion, G.; Lopez, J.P.; Navarro-Sanchez, L.; Yerko, V.; Mechawar, N.; Szyf, M.; Meaney, M.J.; Turecki, G. Genome-wide methylation changes in the brains of suicide completers. Am. J. Psychiatry 2013, 170, 511–520. [Google Scholar] [CrossRef] [PubMed]
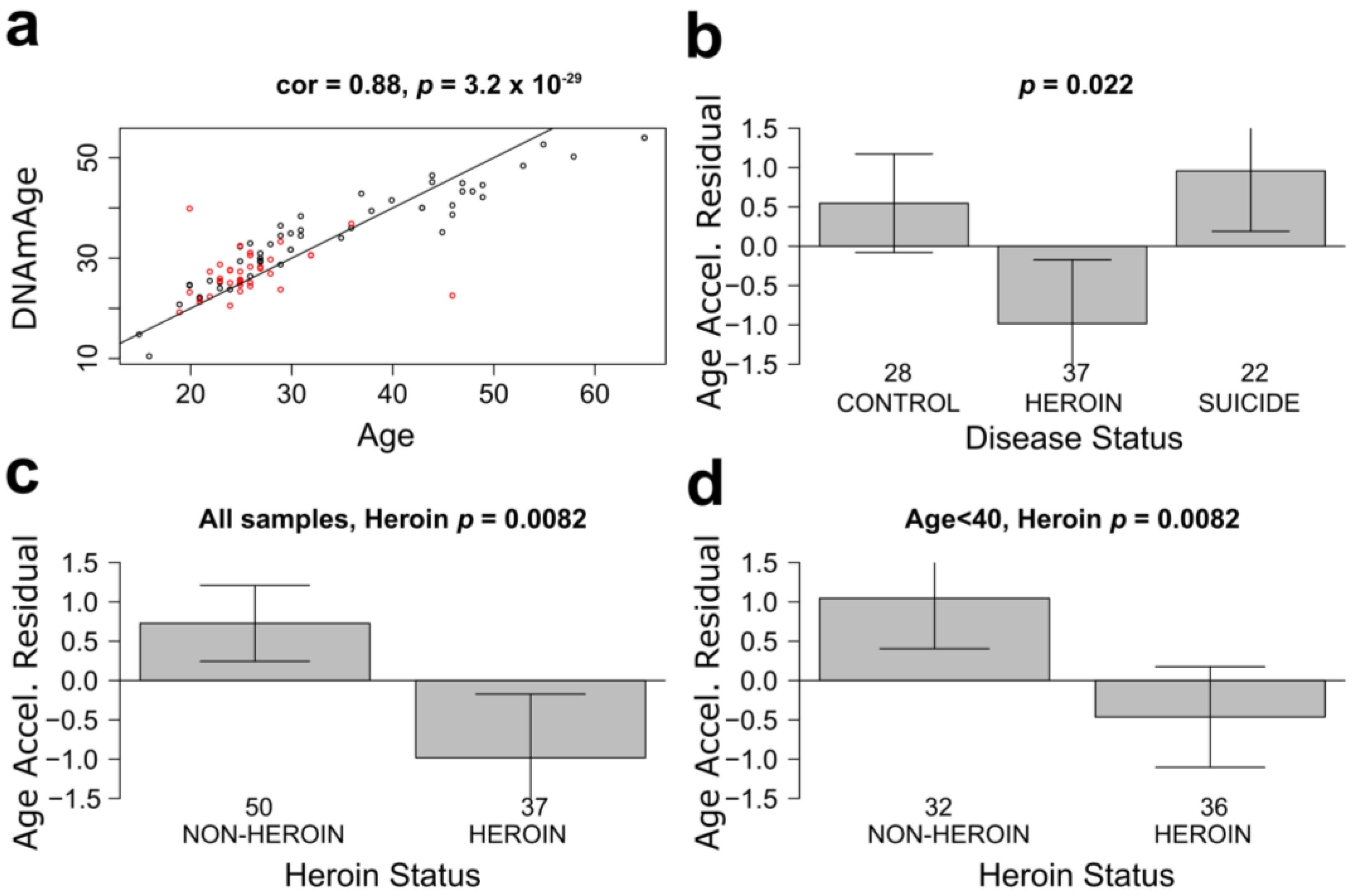

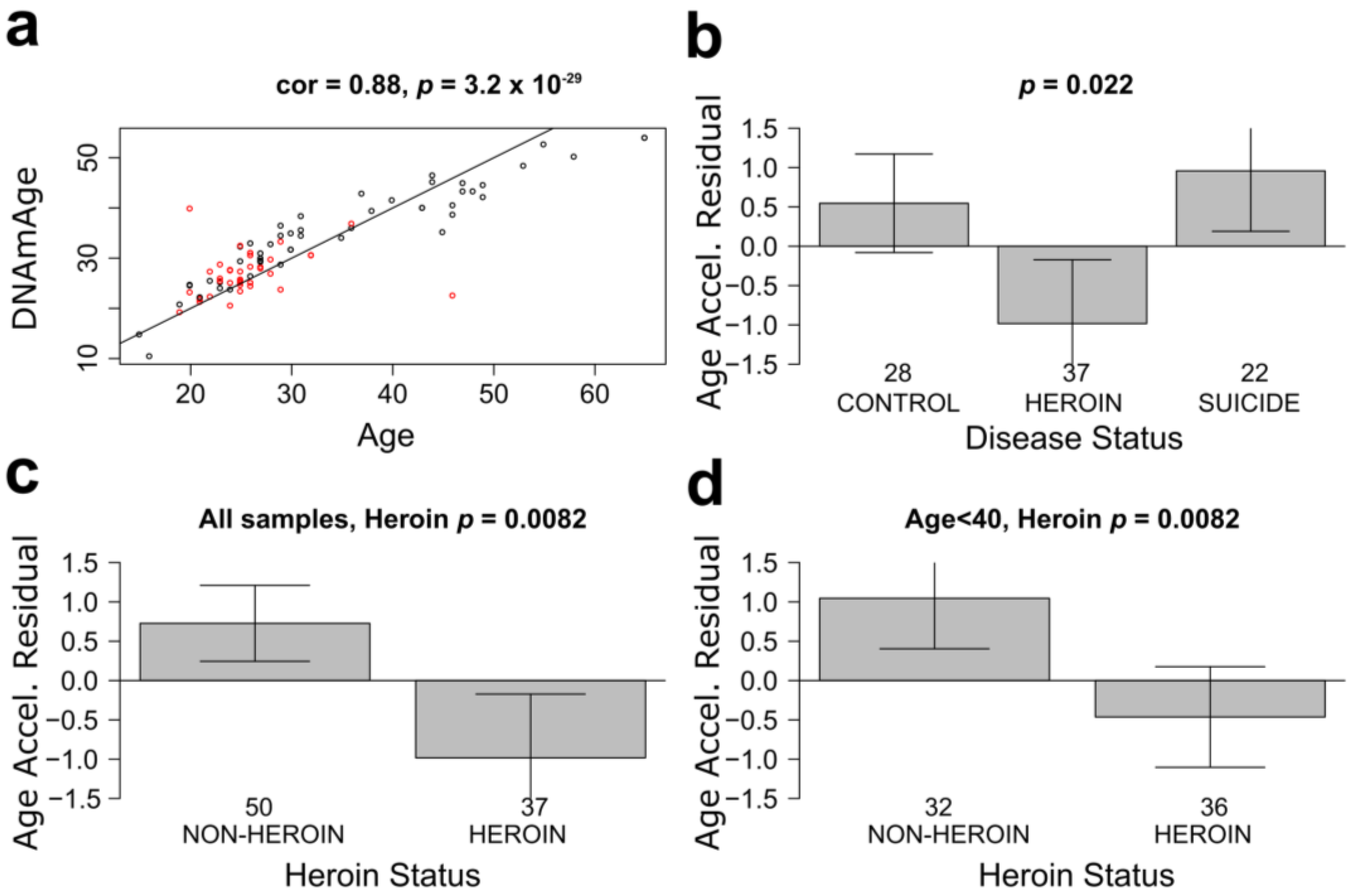{kind=link}
| Genomic Features/Putative Enhancers | Heroin DMSs | Suicide DMSs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HyperDMSs | HypoDMSs | HyperDMSs | HypoDMSs | ||||||
| Odds Ratio | p-Value | Odds Ratio | p-Value | Odds Ratio | p-Value | Odds Ratio | p-Value | ||
| Genic features | Exons | 1.41 | 1.6 × 10−4 | 0.85 | 0.19 | 1.36 | 0.045 | 0.85 | 0.54 |
| Introns | 1.21 | 0.013 | 0.97 | 0.75 | 1.31 | 0.030 | 1.15 | 0.38 | |
| Gene Body | 1.26 | 0.0025 | 1.06 | 0.52 | 1.29 | 0.044 | 0.94 | 0.70 | |
| Intergenic | 1.11 | 0.20 | 0.86 | 0.15 | 0.95 | 0.78 | 1.14 | 0.43 | |
| Promoters | 0.47 | 4.8 × 10−15 | 1.32 | 0.0034 | 0.53 | 6.1 × 10−5 | 0.83 | 0.34 | |
| H3K27ac ChIP-seq peaks (human PFC) | Distal H3K27ac peaks (enhancers) | 1.18 | 0.07017 | 1.52 | 3.0 × 10−5 | 0.94 | 0.76 | 1.11 | 0.57 |
| Proximal H3K27ac peaks (promoters) | 0.33 | 2.0 × 10−15 | 1.52 | 1.2 × 10−4 | 0.67 | 0.048 | 0.60 | 0.052 | |
| CpG island-related features | CpG islands | 1.18 | 0.03 | 1.02 | 0.85 | 0.91 | 0.52 | 0.86 | 0.42 |
| Shores | 1.16 | 0.075 | 1.22 | 0.044 | 0.86 | 0.42 | 0.85 | 0.29 | |
| Other regions | 0.76 | 3.5 × 10−4 | 0.85 | 0.06 | 1.22 | 0.12 | 1.19 | 0.26 | |
| DM/DE Genes | Glu-DE Genes | N = 1074 Genes | GABA-DE Genes | N = 701 Genes | |
|---|---|---|---|---|---|
| Overlapping | p-Value | Overlapping | p-Value | ||
| hyperDMGs | N = 537 genes | ||||
| heroin | 70 | 2.2 × 10−5 | 27 | 0.59 | |
| hypoDMGs | N = 405 genes | ||||
| heroin | 40 | 0.087 | 31 | 0.019 | |
| hyperDMGs | N = 201 genes | ||||
| suicide | 34 | 1.9 × 10−5 | 11 | 0.47 | |
| hypoDMGs | N = 133 genes | ||||
| suicide | 14 | 0.17 | 15 | 3.6 × 10−3 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlenkov, A.; Jaffe, A.E.; Timashpolsky, A.; Apontes, P.; Rudchenko, S.; Barbu, M.; Byne, W.; Hurd, Y.L.; Horvath, S.; Dracheva, S. DNA Methylation Profiling of Human Prefrontal Cortex Neurons in Heroin Users Shows Significant Difference between Genomic Contexts of Hyper- and Hypomethylation and a Younger Epigenetic Age. Genes 2017, 8, 152. https://doi.org/10.3390/genes8060152
Kozlenkov A, Jaffe AE, Timashpolsky A, Apontes P, Rudchenko S, Barbu M, Byne W, Hurd YL, Horvath S, Dracheva S. DNA Methylation Profiling of Human Prefrontal Cortex Neurons in Heroin Users Shows Significant Difference between Genomic Contexts of Hyper- and Hypomethylation and a Younger Epigenetic Age. Genes. 2017; 8(6):152. https://doi.org/10.3390/genes8060152
Chicago/Turabian StyleKozlenkov, Alexey, Andrew E. Jaffe, Alisa Timashpolsky, Pasha Apontes, Sergei Rudchenko, Mihaela Barbu, William Byne, Yasmin L. Hurd, Steve Horvath, and Stella Dracheva. 2017. "DNA Methylation Profiling of Human Prefrontal Cortex Neurons in Heroin Users Shows Significant Difference between Genomic Contexts of Hyper- and Hypomethylation and a Younger Epigenetic Age" Genes 8, no. 6: 152. https://doi.org/10.3390/genes8060152
APA StyleKozlenkov, A., Jaffe, A. E., Timashpolsky, A., Apontes, P., Rudchenko, S., Barbu, M., Byne, W., Hurd, Y. L., Horvath, S., & Dracheva, S. (2017). DNA Methylation Profiling of Human Prefrontal Cortex Neurons in Heroin Users Shows Significant Difference between Genomic Contexts of Hyper- and Hypomethylation and a Younger Epigenetic Age. Genes, 8(6), 152. https://doi.org/10.3390/genes8060152