
Deoxynucleoside Salvage in Fission Yeast Allows Rescue of Ribonucleotide Reductase Deficiency but Not Spd1-Mediated Inhibition of Replication

 , , , , ,
, , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular and Genetic Procedures
2.2. Physiological Experiments and Cell Biology
3. Results
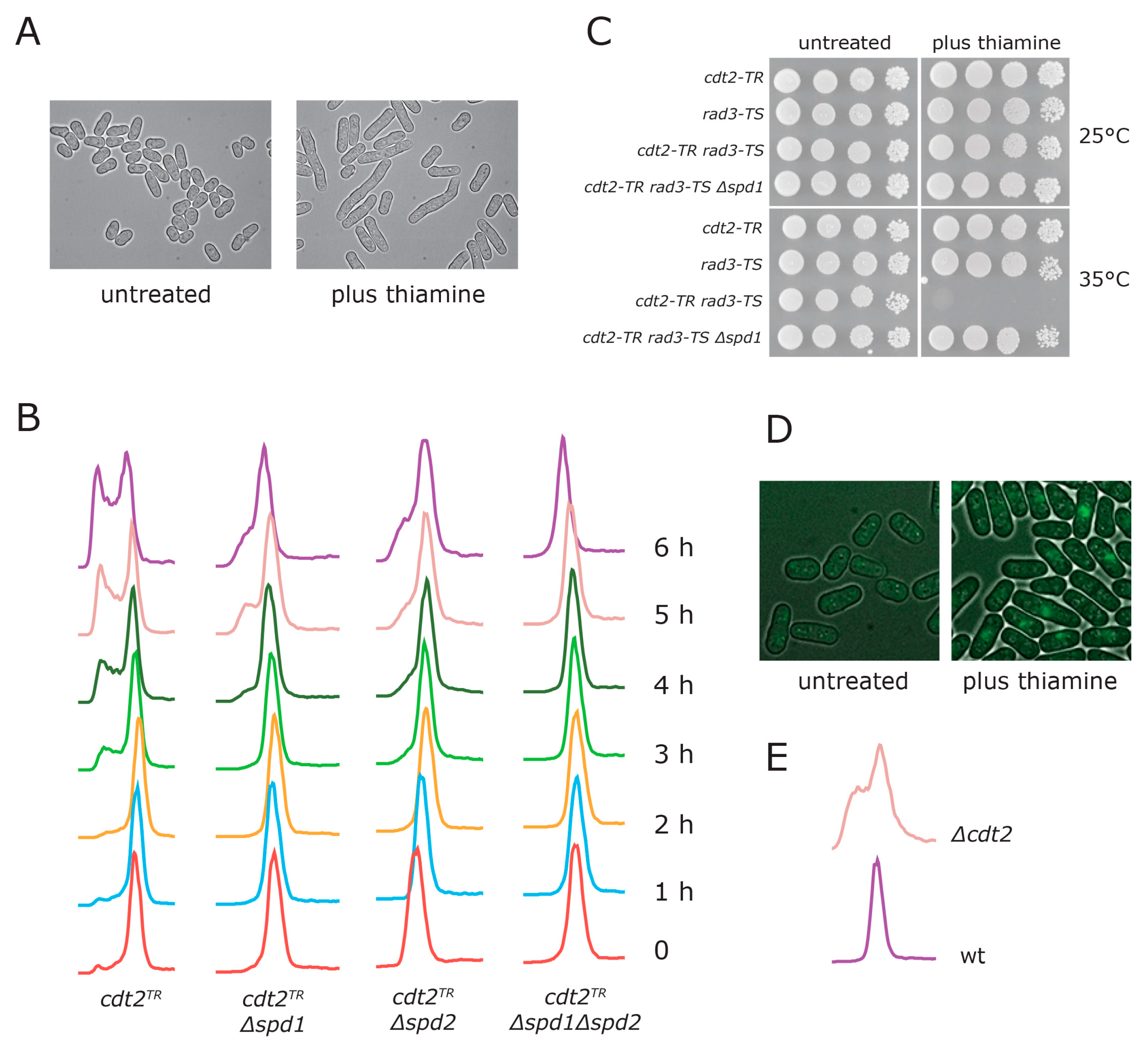
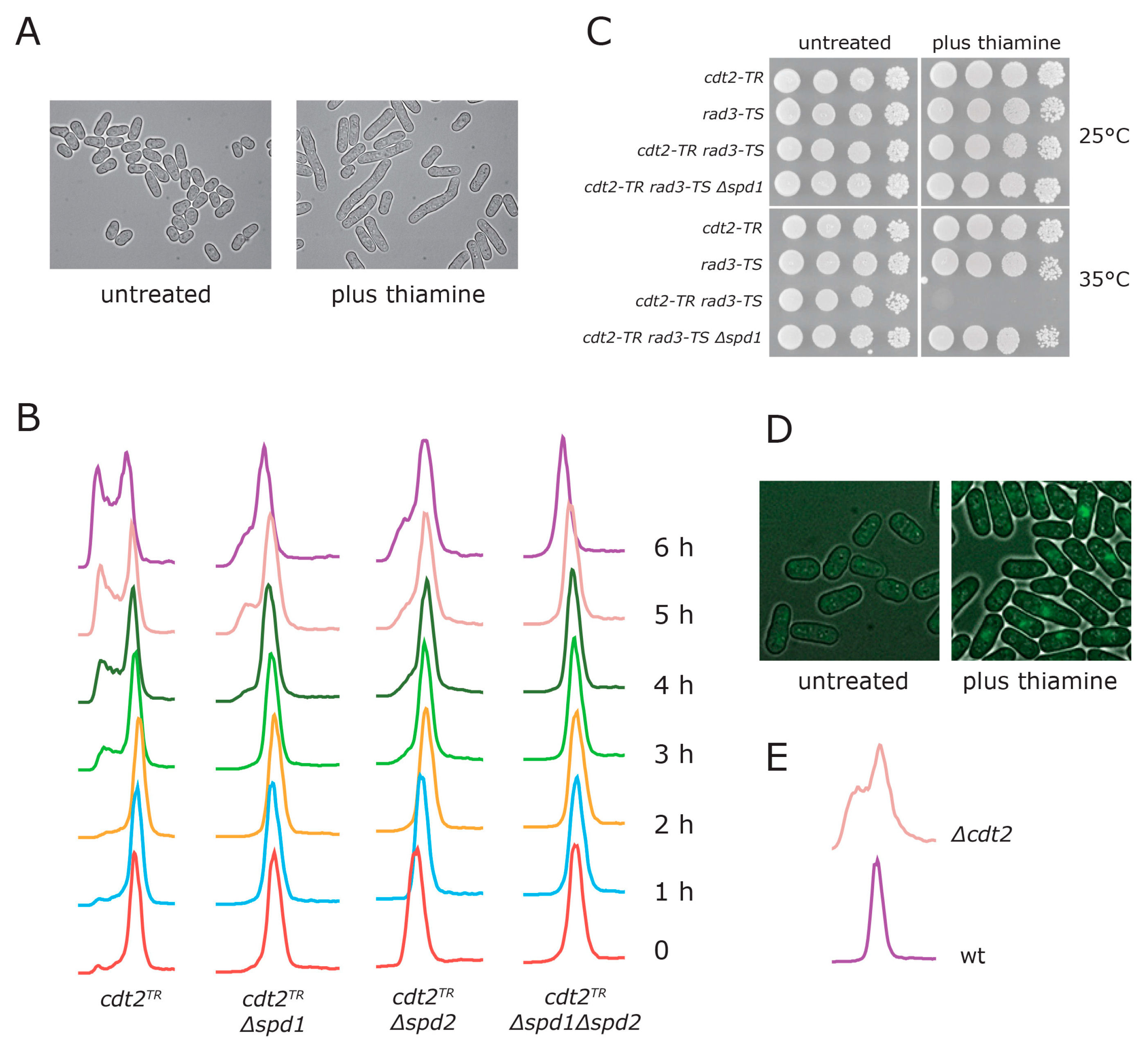
3.1. Generation of a Conditional CRL4Cdt2 Mutant
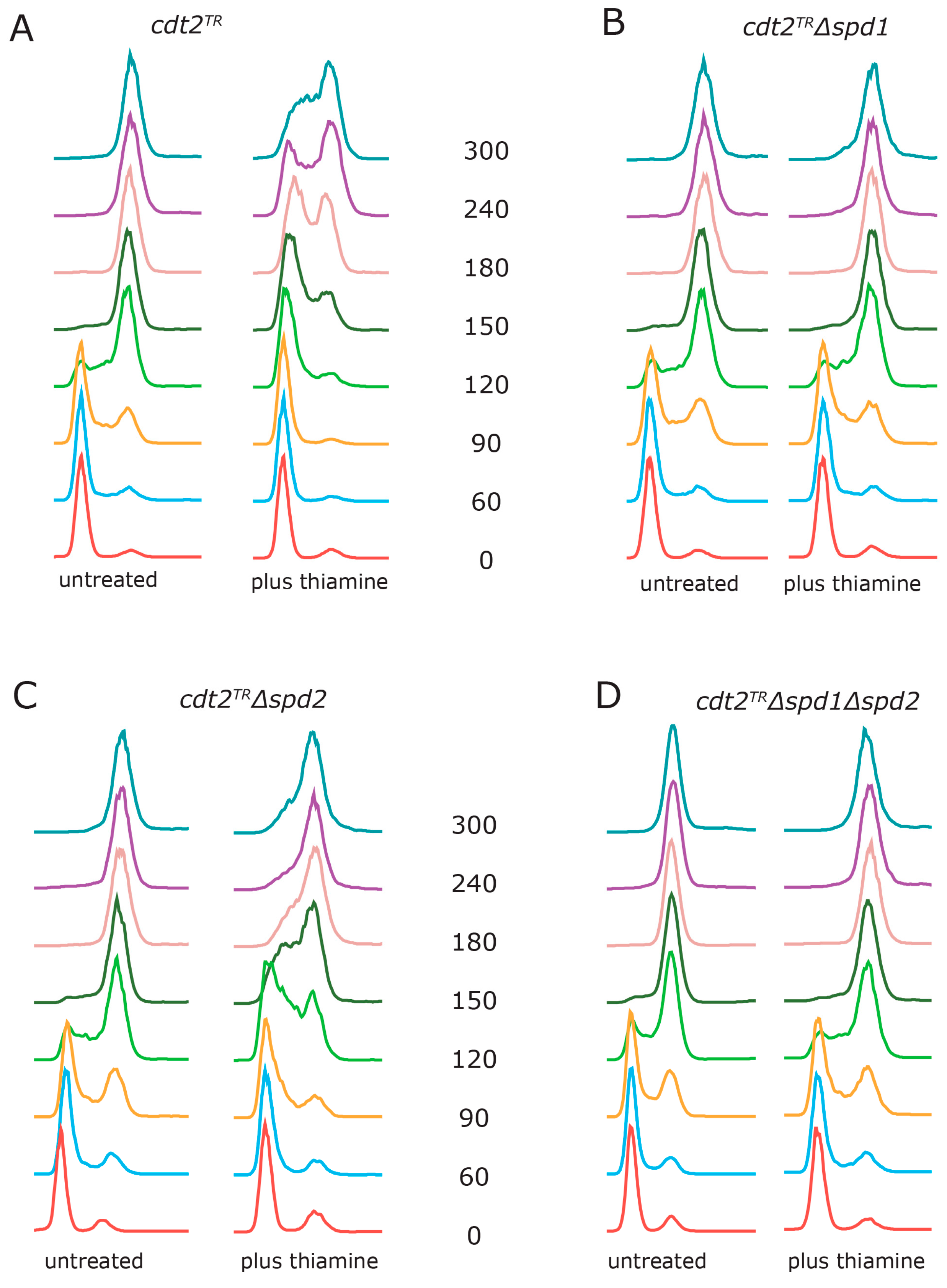
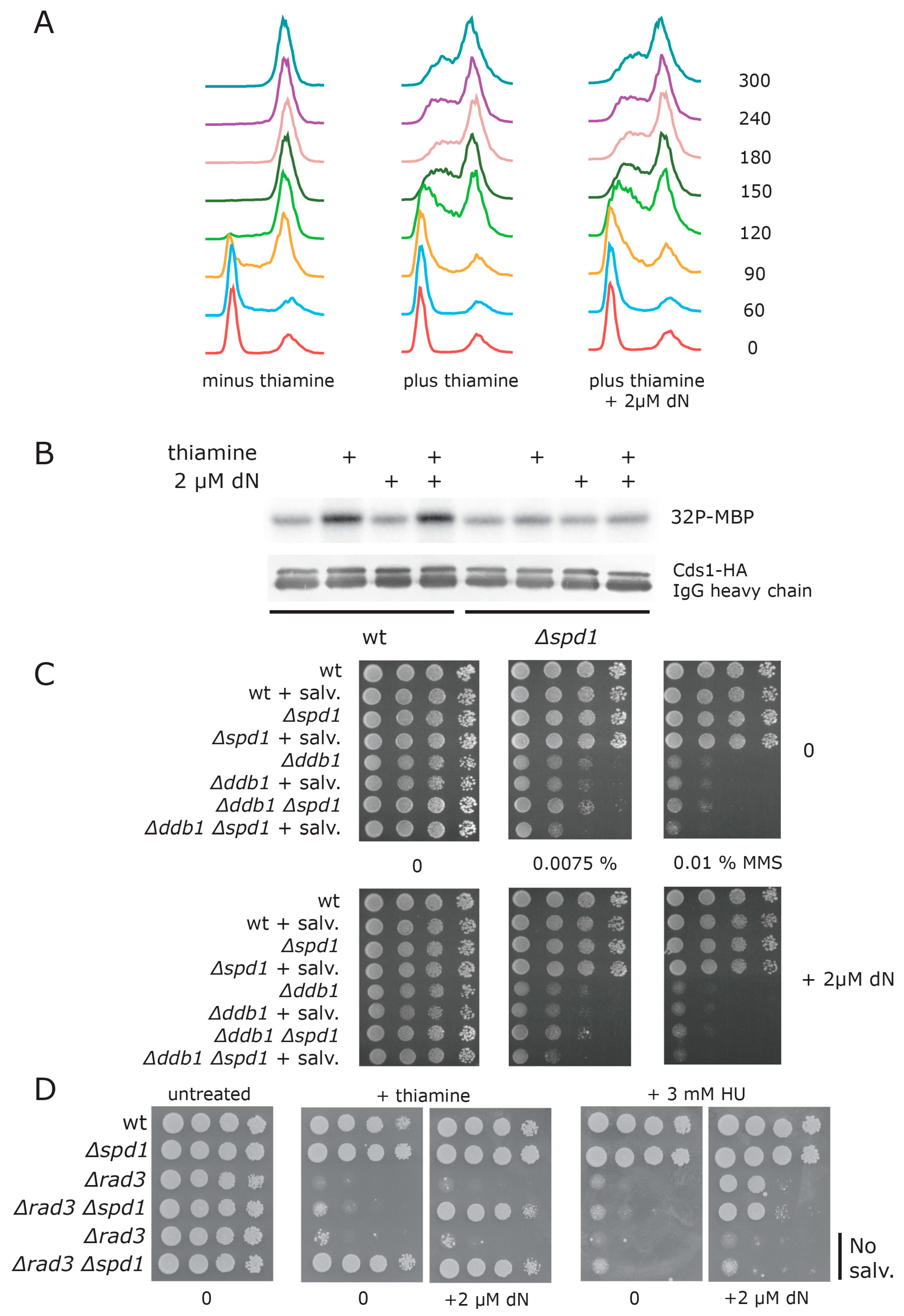
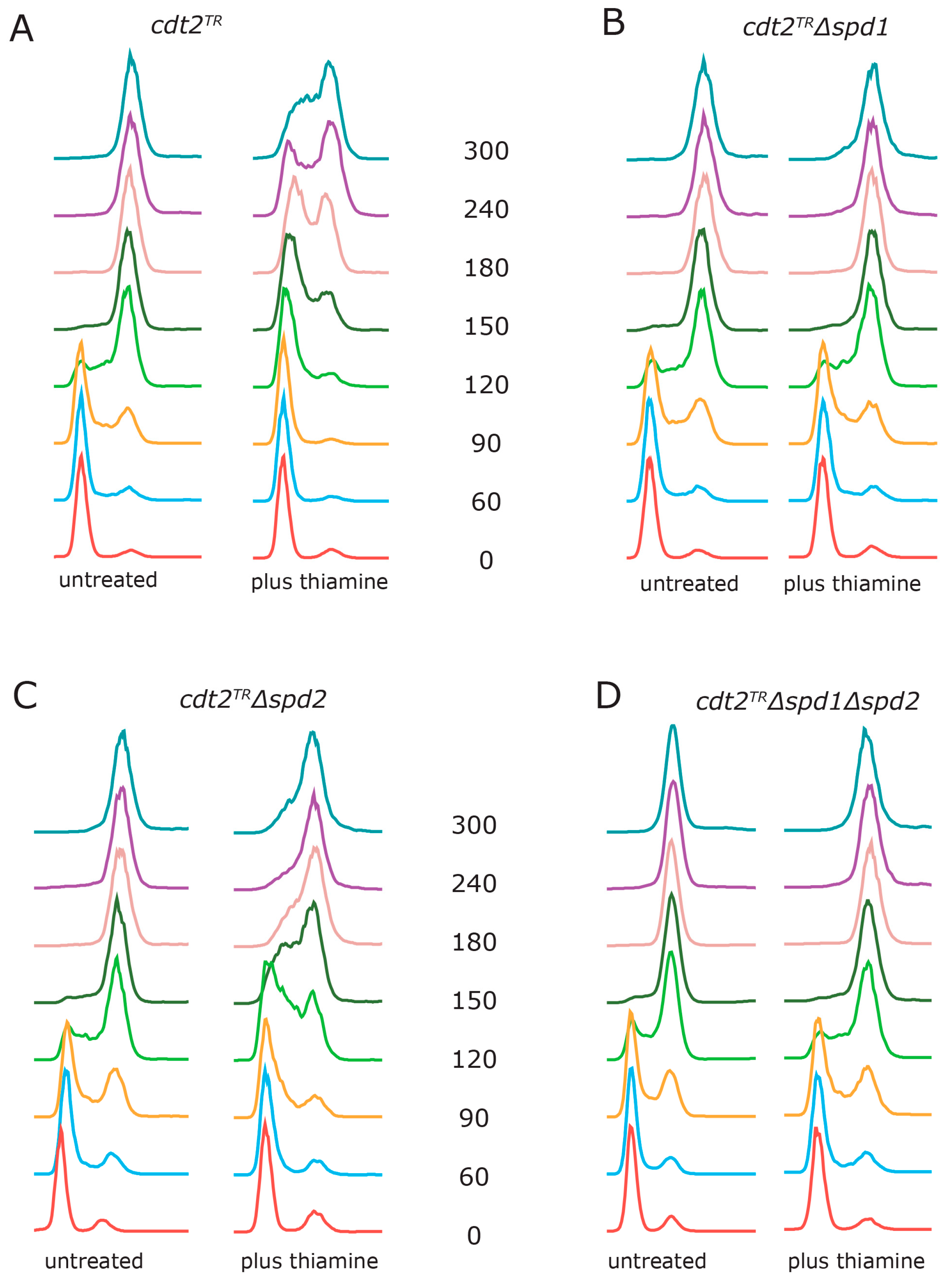
3.2. Spd1 Accumulation Causes S Phase Delay
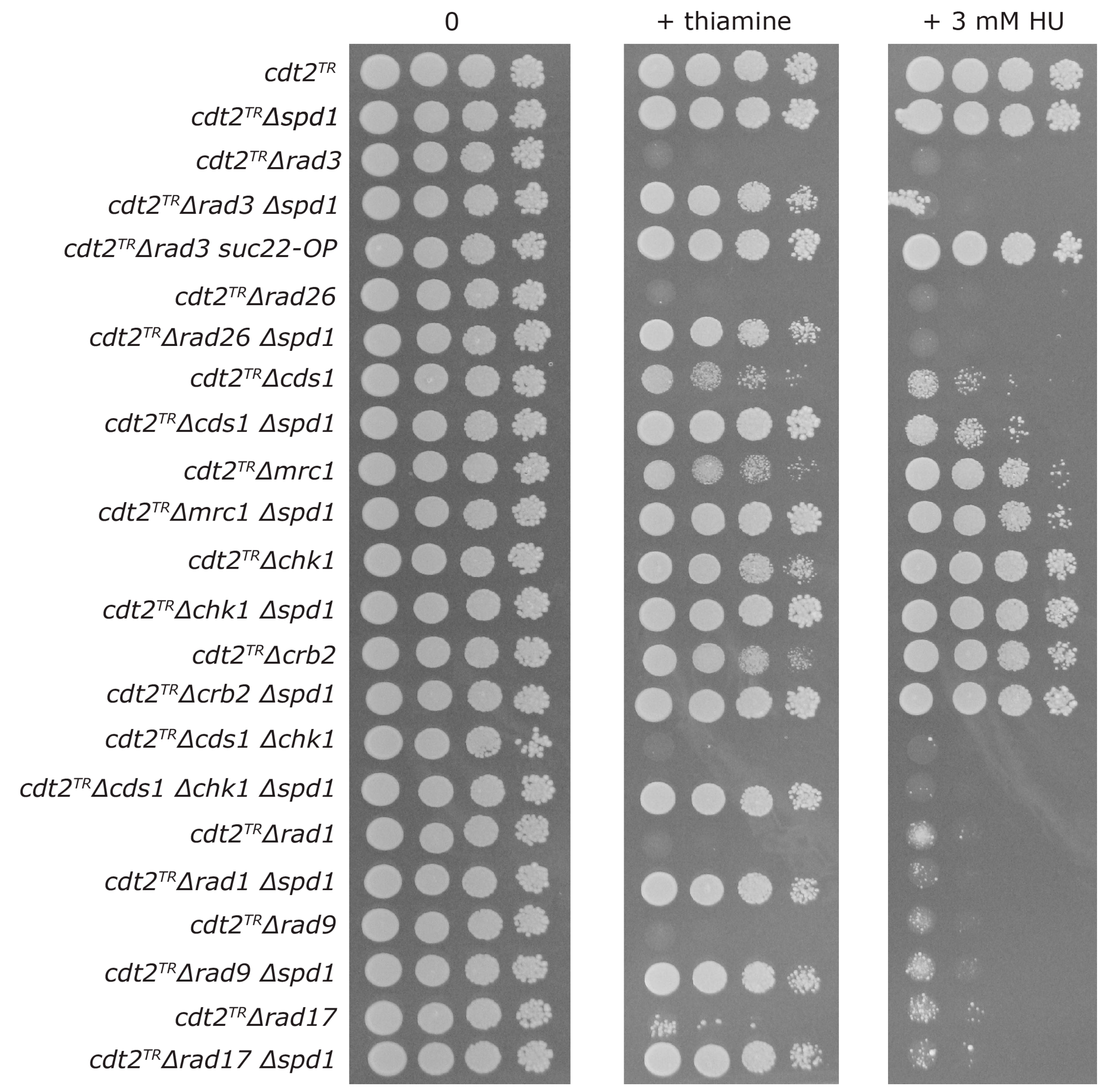
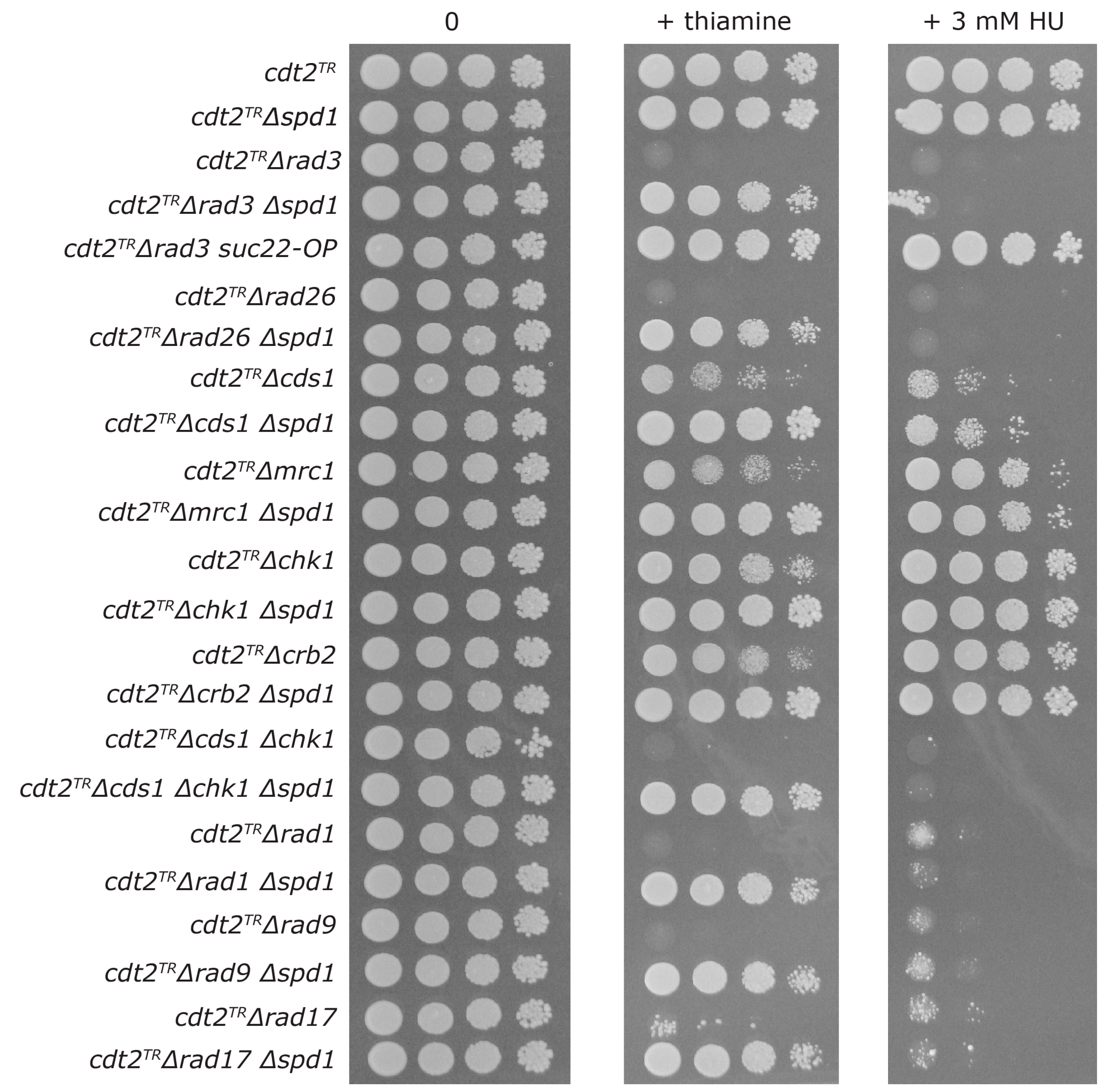
3.3. Both Branches of the Rad3 Checkpoint Are Involved in Tolerating Replication Problems Caused by Spd1 Accumulation
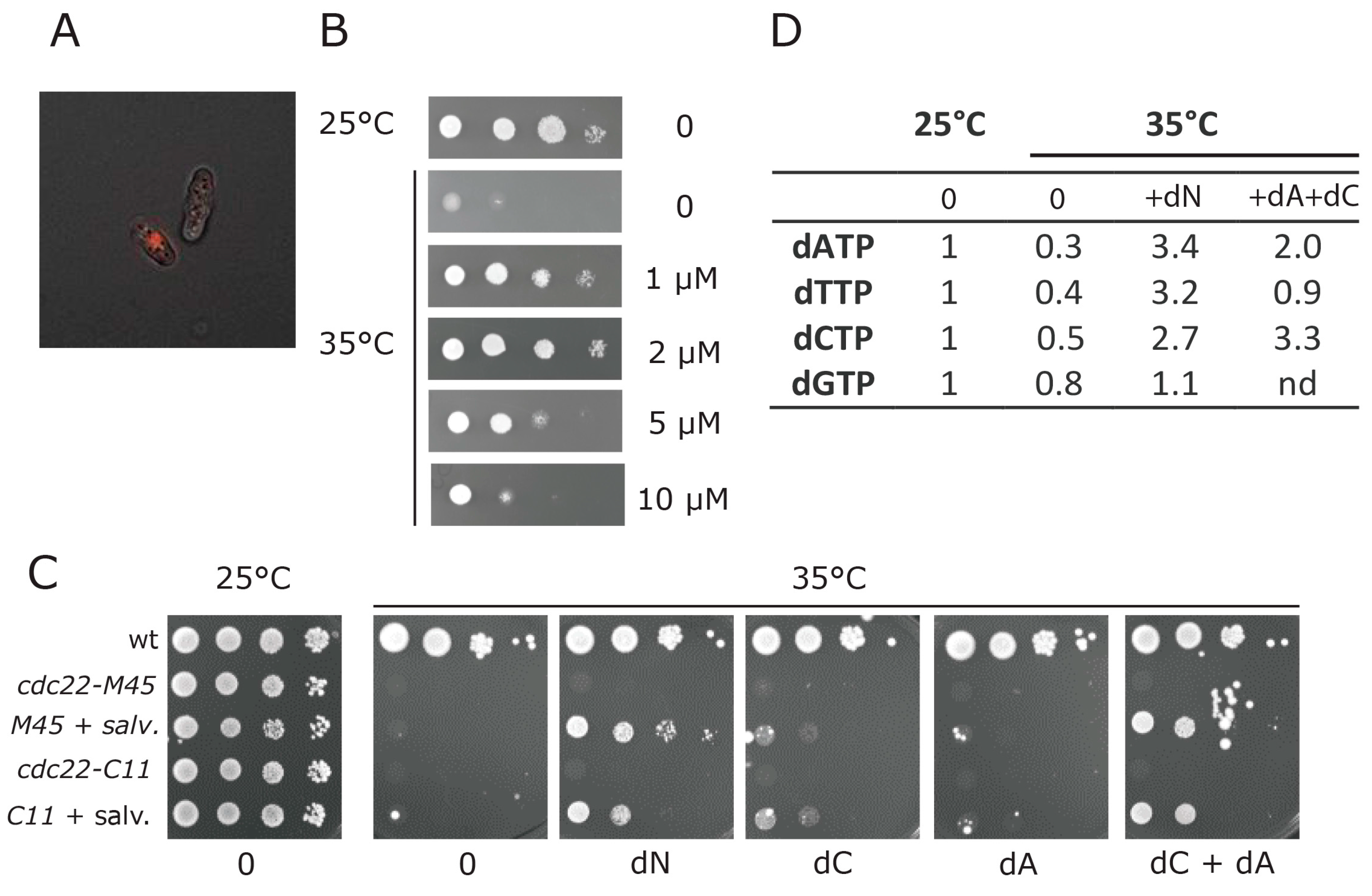
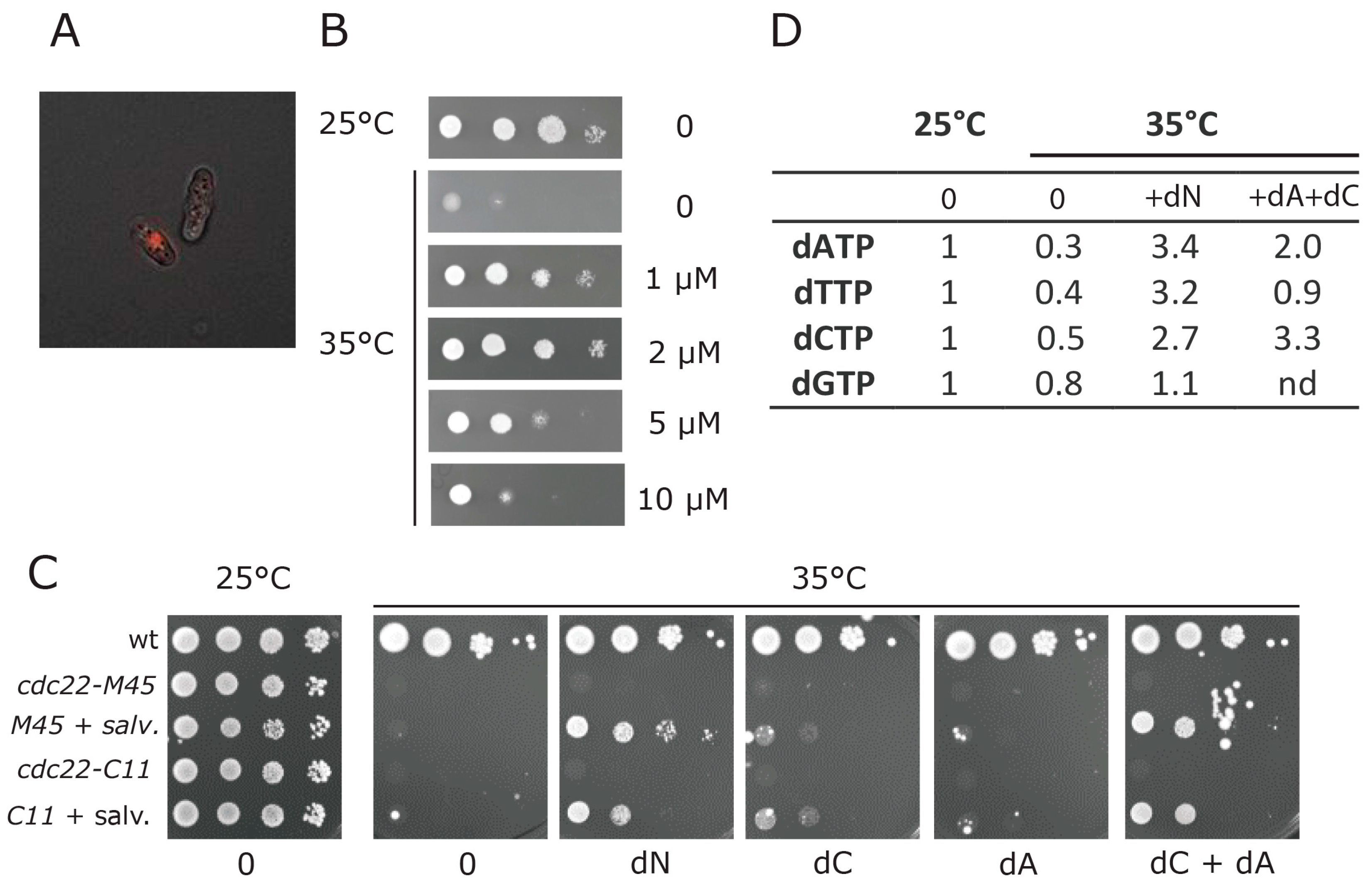
3.4. Establishment of a Deoxynucleotide Salvage Pathway in Fission Yeast
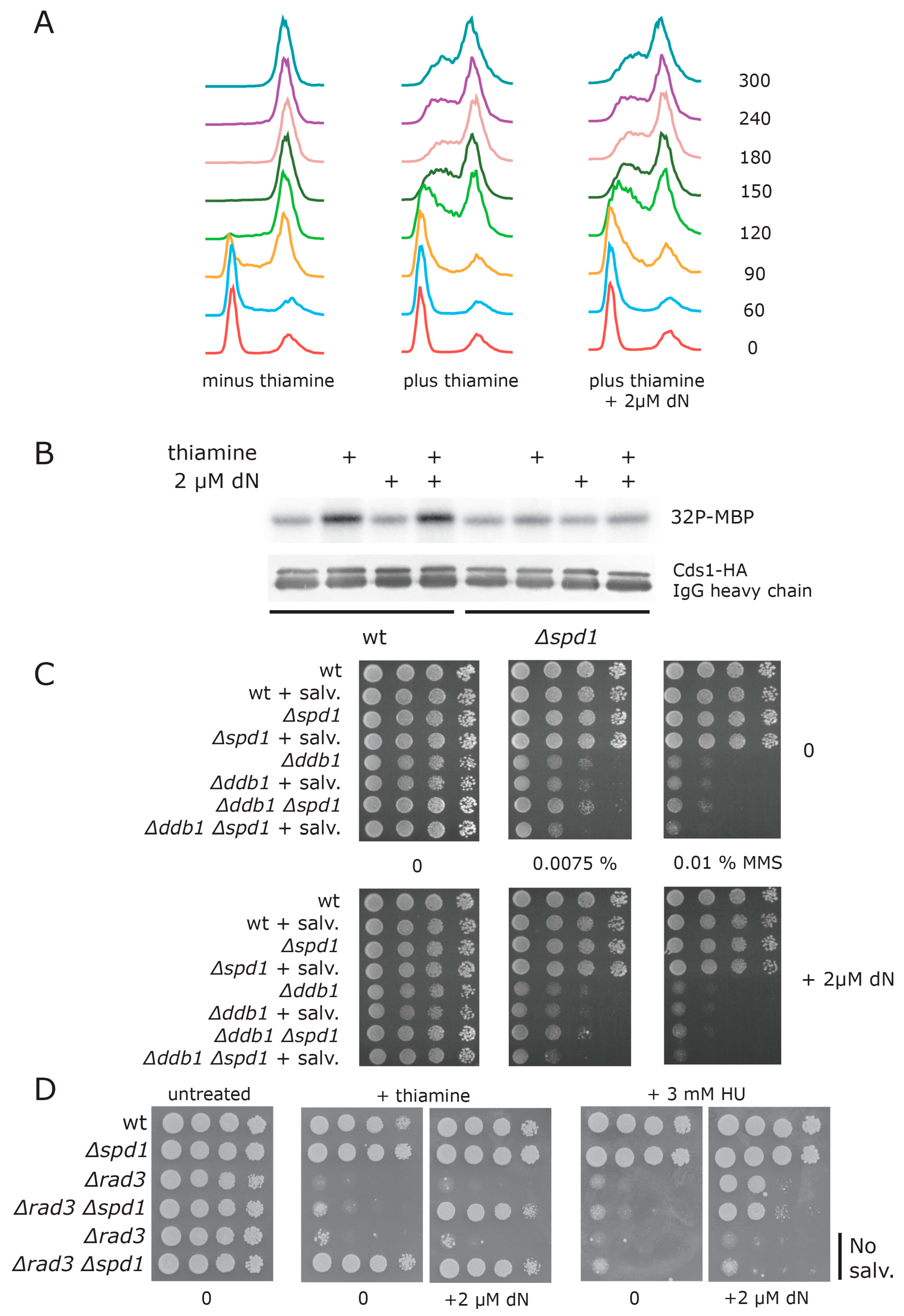
3.5. dNTP Salvage Does Not Rescue Spd1 Accumulation
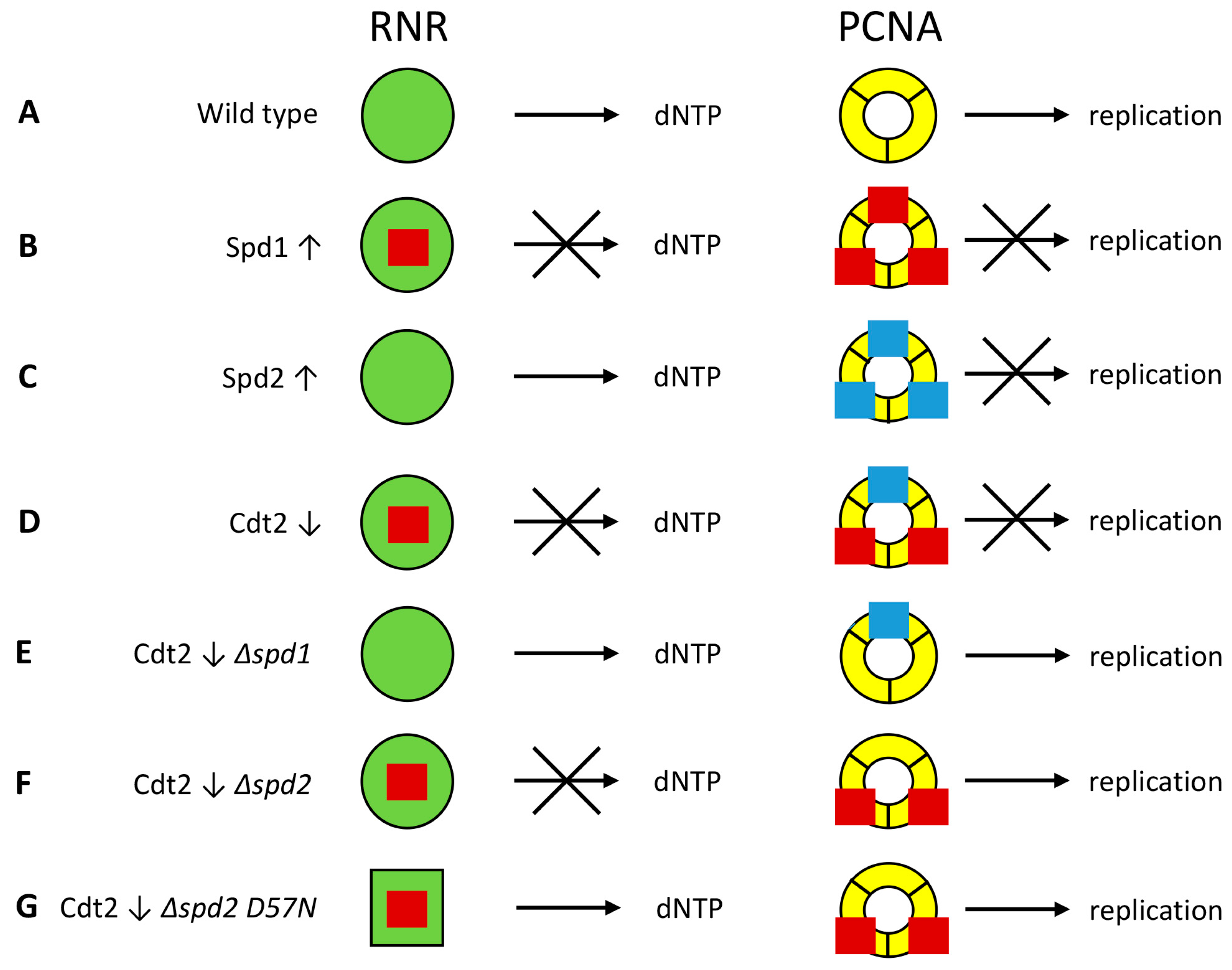
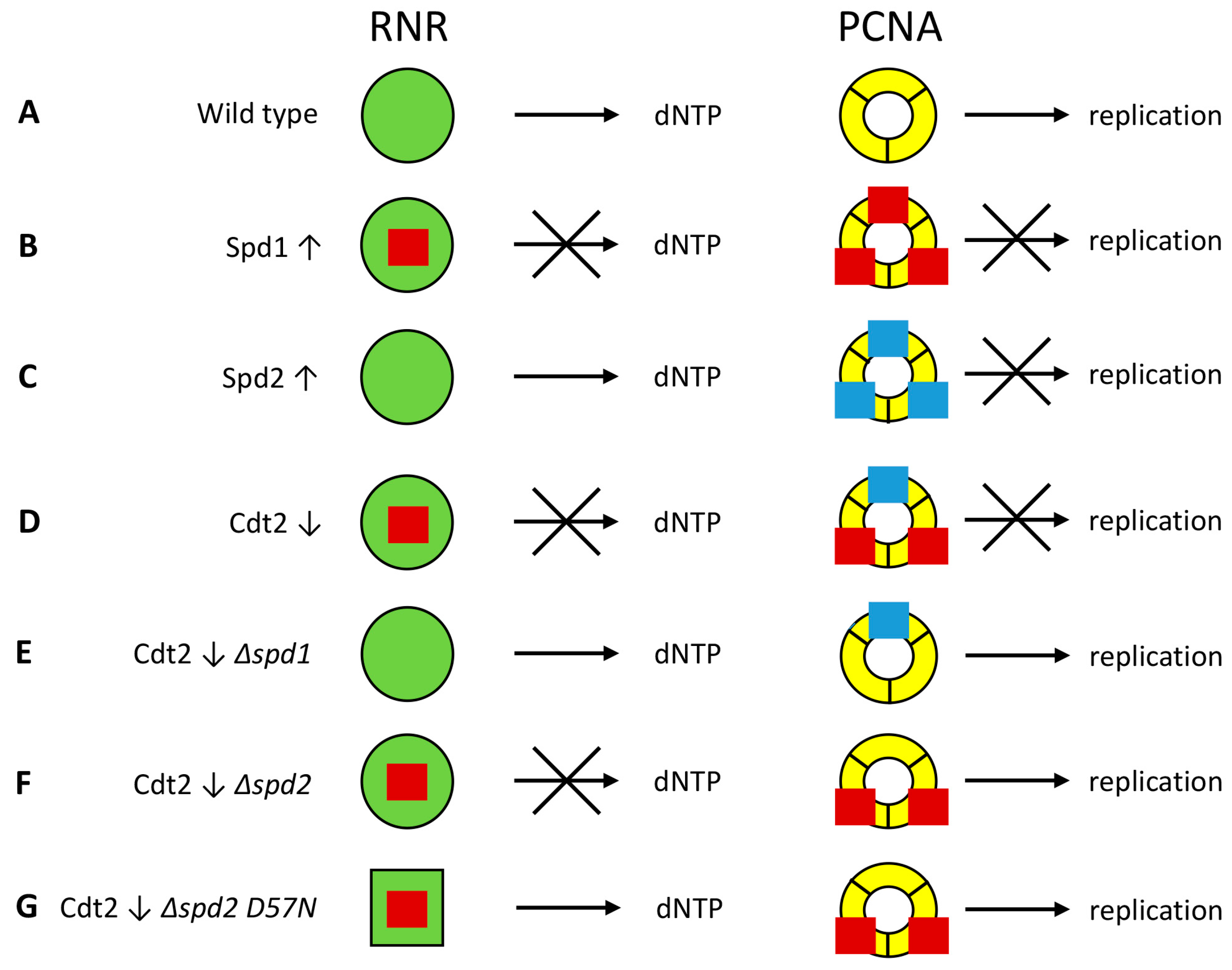
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell. 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Stodola, J.L.; Burgers, P.M. Resolving individual steps of Okazaki-fragment maturation at a millisecond timescale. Nat. Struct. Mol. Biol. 2016, 23, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Walter, J.C. Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011, 25, 1568–1582. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Powell, K.A.; Mundt, K.; Wu, L.; Carr, A.M.; Caspari, T. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 2003, 17, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Salguero, I.; Guarino, E.; Shepherd, M.E.; Deegan, T.D.; Havens, C.G.; MacNeill, S.A.; Walter, J.C.; Kearsey, S.E. Ribonucleotide reductase activity is coupled to DNA synthesis via proliferating cell nuclear antigen. Curr. Biol. 2012, 22, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, C.; Fleck, O.; Hansen, H.A.; Liu, C.; Slaaby, R.; Carr, A.M.; Nielsen, O. Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 2005, 19, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Nestoras, K.; Mohammed, A.H.; Schreurs, A.S.; Fleck, O.; Watson, A.T.; Poitelea, M.; O’Shea, C.; Chahwan, C.; Holmberg, C.; Kragelund, B.B.; et al. Regulation of ribonucleotide reductase by Spd1 involves multiple mechanisms. Genes Dev. 2010, 24, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Poitelea, M.; Watson, A.; Yoshida, S.H.; Shimoda, C.; Holmberg, C.; Nielsen, O.; Carr, A.M. Transactivation of Schizosaccharomyces pombe cdt2+ stimulates a Pcu4-Ddb1-CSN ubiquitin ligase. EMBO J. 2005, 24, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, C.; Nielsen, O. Replication: DNA building block synthesis on demand. Curr. Biol. 2012, 22, R271–R272. [Google Scholar] [CrossRef] [PubMed]
- Woollard, A.; Basi, G.; Nurse, P. A novel S phase inhibitor in fission yeast. EMBO J. 1996, 15, 4603–4612. [Google Scholar] [PubMed]
- Moss, J.; Tinline-Purvis, H.; Walker, C.A.; Folkes, L.K.; Stratford, M.R.; Hayles, J.; Hoe, K.L.; Kim, D.U.; Park, H.O.; Kearsey, S.E.; et al. Break-induced ATR and Ddb1-Cul4Cdt2 ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 2010, 24, 2705–2716. [Google Scholar] [CrossRef] [PubMed]
- Fleck, O.; Vejrup-Hansen, R.; Watson, A.; Carr, A.M.; Nielsen, O.; Holmberg, C. Spd1 accumulation causes genome instability independently of ribonucleotide reductase activity but functions to protect the genome when deoxynucleotide pools are elevated. J. Cell Sci. 2013, 126, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, P.; Dahl, L.; Chilkova, O.; Domkin, V.; Thelander, L. The Schizosaccharomyces pombe replication inhibitor Spd1 regulates ribonucleotide reductase activity and dNTPs by binding to the large Cdc22 subunit. J. Biol. Chem. 2006, 281, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.H.; Al-Amodi, H.; Nakamura, T.; McInerny, C.J.; Shimoda, C. The Schizosaccharomyces pombe cdt2+ gene, a target of G1-S phase-specific transcription factor complex DSC1, is required for mitotic and premeiotic DNA replication. Genetics 2003, 164, 881–893. [Google Scholar] [PubMed]
- Vejrup-Hansen, R.; Fleck, O.; Landvad, K.; Fahnoe, U.; Broendum, S.S.; Schreurs, A.S.; Kragelund, B.B.; Carr, A.M.; Holmberg, C.; Nielsen, O. Spd2 assists Spd1 in the modulation of ribonucleotide reductase architecture but does not regulate deoxynucleotide pools. J. Cell Sci. 2014, 127, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Klar, A.; Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991, 194, 795–823. [Google Scholar] [PubMed]
- Matsuyama, A.; Shirai, A.; Yashiroda, Y.; Kamata, A.; Horinouchi, S.; Yoshida, M. pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast 2004, 21, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Vernis, L.; Piskur, J.; Diffley, J.F. Reconstitution of an efficient thymidine salvage pathway in Saccharomyces cerevisiae. Nucleic Acids Res. 2003, 31, e120. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Porter-Goff, M.; Patel, P.K.; Benoit, K.; Rhind, N. In vivo labeling of fission yeast DNA with thymidine and thymidine analogs. Methods 2004, 33, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Egel, R.; Willer, M.; Kjaerulff, S.; Davey, J.; Nielsen, O. Assessment of pheromone production and response in fission yeast by a halo test of induced sporulation. Yeast 1994, 10, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O. Synchronization of S Phase in Schizosaccharomyces pombe Cells by Transient Exposure to M-Factor Pheromone. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kearsey, S.E. Monitoring DNA replication in fission yeast by incorporation of 5-ethynyl-2’-deoxyuridine. Nucleic Acids Res. 2011, 39, e60. [Google Scholar] [CrossRef] [PubMed]
- Basi, G.; Schmid, E.; Maundrell, K. TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene 1993, 123, 131–136. [Google Scholar] [CrossRef]
- Munch-Petersen, B.; Knecht, W.; Lenz, C.; Sondergaard, L.; Piskur, J. Functional expression of a multisubstrate deoxyribonucleoside kinase from Drosophila melanogaster and its C-terminal deletion mutants. J. Biol. Chem. 2000, 275, 6673–6679. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.B.; Fantes, P.A. The cdc22 gene of Schizosaccharomyces pombe encodes a cell cycle-regulated transcript. EMBO J. 1986, 5, 2981–2985. [Google Scholar] [PubMed]
- Lindsay, H.D.; Griffiths, D.J.; Edwards, R.J.; Christensen, P.U.; Murray, J.M.; Osman, F.; Walworth, N.; Carr, A.M. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998, 12, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Munch-Petersen, B.; Piskur, J.; Sondergaard, L. Four deoxynucleoside kinase activities from Drosophila melanogaster are contained within a single monomeric enzyme, a new multifunctional deoxynucleoside kinase. J. Biol. Chem. 1998, 273, 3926–3931. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Lemke, E.A. Mapping multivalency and differential affinities within large intrinsically disordered protein complexes with segmental motion analysis. Angew. Chem. 2014, 53, 7364–7367. [Google Scholar] [CrossRef] [PubMed]
- Tournier, S.; Leroy, D.; Goubin, F.; Ducommun, B.; Hyams, J.S. Heterologous expression of the human cyclin-dependent kinase inhibitor p21Cip1 in the fission yeast, Schizosaccharomyces pombe reveals a role for PCNA in the chk1+ cell cycle checkpoint pathway. Mol. Biol. Cell 1996, 7, 651–662. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleck, O.; Fahnøe, U.; Løvschal, K.V.; Gasasira, M.-F.U.; Marinova, I.N.; Kragelund, B.B.; Carr, A.M.; Hartsuiker, E.; Holmberg, C.; Nielsen, O. Deoxynucleoside Salvage in Fission Yeast Allows Rescue of Ribonucleotide Reductase Deficiency but Not Spd1-Mediated Inhibition of Replication. Genes 2017, 8, 128. https://doi.org/10.3390/genes8050128
Fleck O, Fahnøe U, Løvschal KV, Gasasira M-FU, Marinova IN, Kragelund BB, Carr AM, Hartsuiker E, Holmberg C, Nielsen O. Deoxynucleoside Salvage in Fission Yeast Allows Rescue of Ribonucleotide Reductase Deficiency but Not Spd1-Mediated Inhibition of Replication. Genes. 2017; 8(5):128. https://doi.org/10.3390/genes8050128
Chicago/Turabian StyleFleck, Oliver, Ulrik Fahnøe, Katrine Vyff Løvschal, Marie-Fabrice Uwamahoro Gasasira, Irina N. Marinova, Birthe B. Kragelund, Antony M. Carr, Edgar Hartsuiker, Christian Holmberg, and Olaf Nielsen. 2017. "Deoxynucleoside Salvage in Fission Yeast Allows Rescue of Ribonucleotide Reductase Deficiency but Not Spd1-Mediated Inhibition of Replication" Genes 8, no. 5: 128. https://doi.org/10.3390/genes8050128
APA StyleFleck, O., Fahnøe, U., Løvschal, K. V., Gasasira, M.-F. U., Marinova, I. N., Kragelund, B. B., Carr, A. M., Hartsuiker, E., Holmberg, C., & Nielsen, O. (2017). Deoxynucleoside Salvage in Fission Yeast Allows Rescue of Ribonucleotide Reductase Deficiency but Not Spd1-Mediated Inhibition of Replication. Genes, 8(5), 128. https://doi.org/10.3390/genes8050128