
Dystrophic Cardiomyopathy—Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cardiomyopathy Associated with Duchenne Muscular Dystrophy
2.1. Overview
2.2. Cellular Pathology of Cardiac Dystrophy
2.2.1. Elevated Intracellular Calcium
Mechanical Damage and Membrane Tears
Stretch-Activated Channels (SACs) and Mechano-Sensitive Transient Receptor Potential (TRP) Channels
Store-Operated Calcium Release
2.2.2. Downstream Consequences of Elevated Intracellular Calcium in the Dystrophic Heart
2.2.3. Functional Muscle Ischemia
2.2.4. Cardiac Mitochondrial Dysfunction
3. Overview of Therapy for DMD-Associated Cardiomyopathy
3.1. Current Therapies
3.1.1. Corticosteroids
3.1.2. ACE Inhibitors and Beta Blockers
3.2. Future Therapies
3.2.1. Membrane Repair
3.2.2. Utrophin Up-Regulation
3.2.3. Stop-Codon Read-Through Therapy
3.2.4. Viral Gene Therapy
3.2.5. Cell-Based Therapy
3.2.6. Endocrine Mediators
3.2.7. Targeting Signalling Pathways
3.2.8. Antisense Oligonucleotides
Overview
2′O-methyl Phosphorothioate AOs
Phosphorodiamidate Morpholino Oligomers
4. Reversal of DMD-Associated Cardiomyopathy Following Treatment with Antisense Oligomers
4.1. Overview
4.2. The Cytoskeletion as a Link between the L-Type Calcium Channel (LTCC) and the Mitochondria
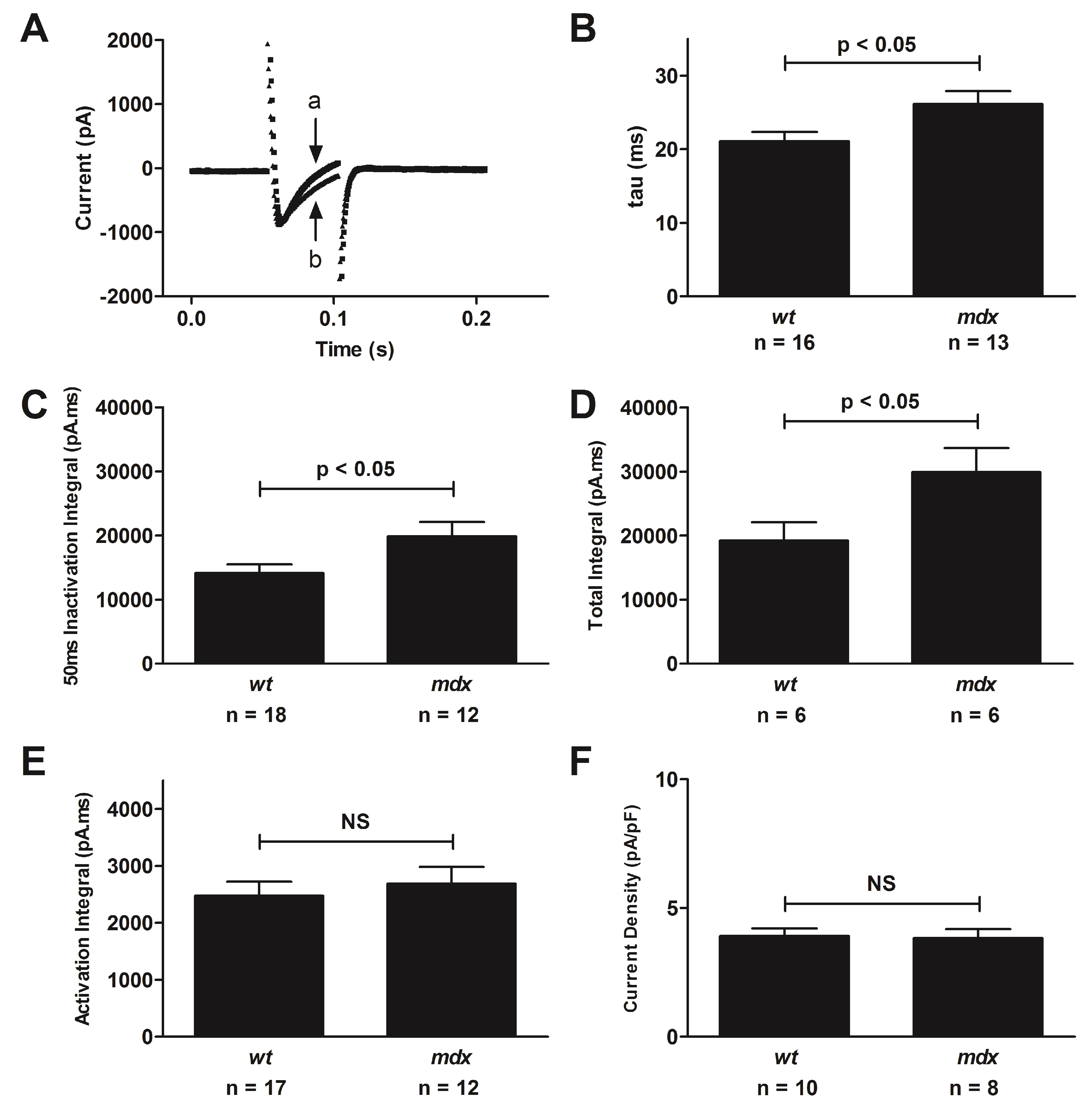
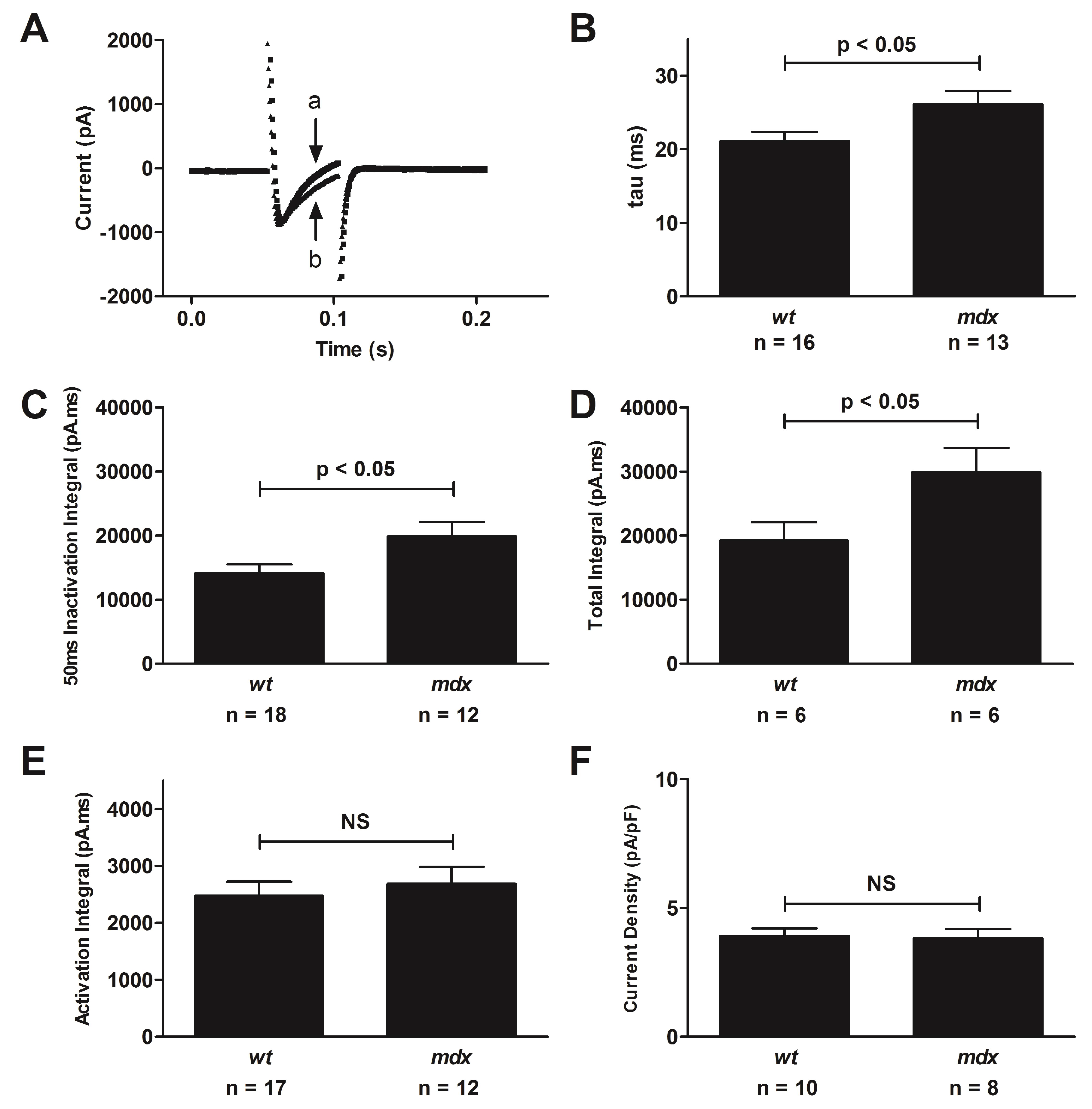
4.3. Altered L-Type Calcium Channel Function in the Mdx Mouse
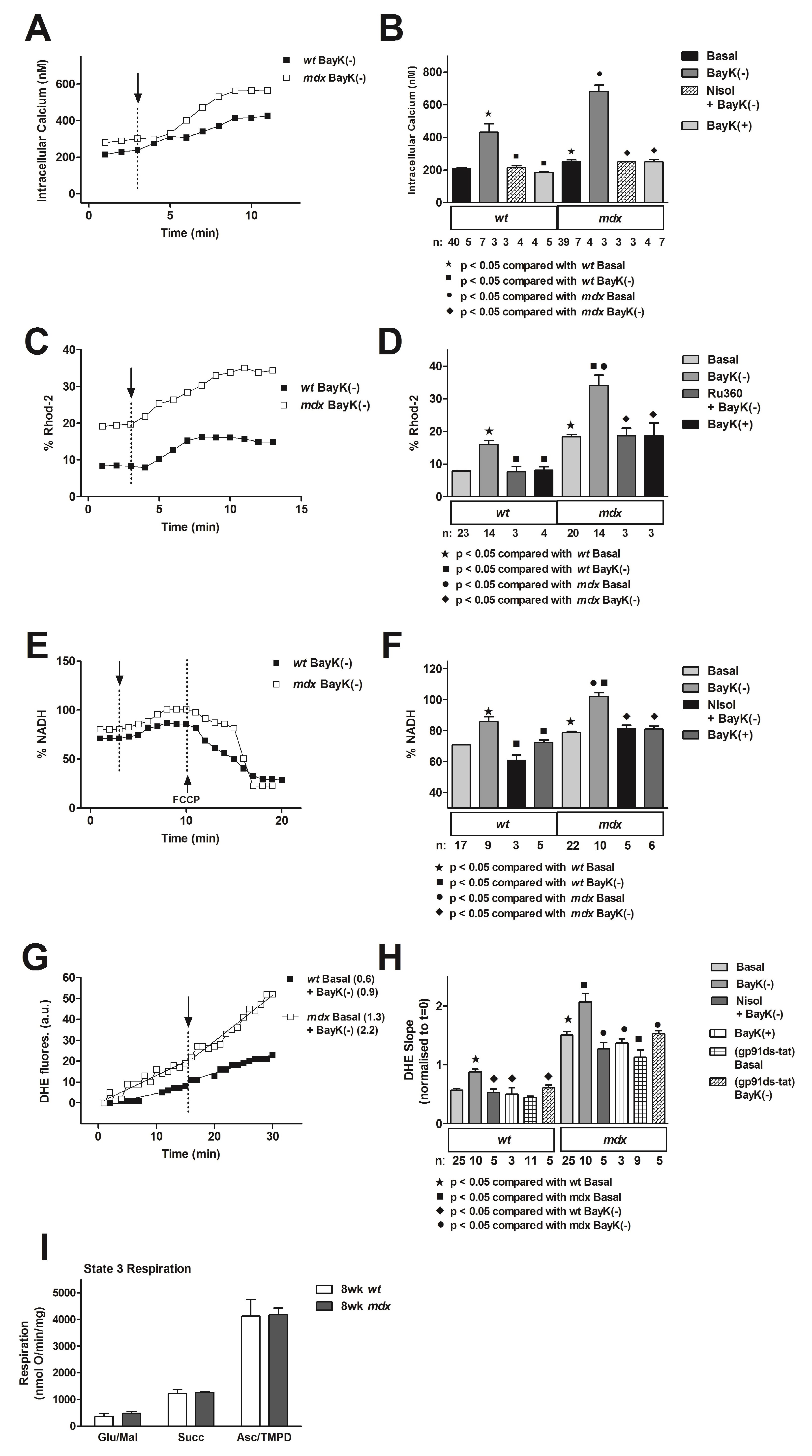
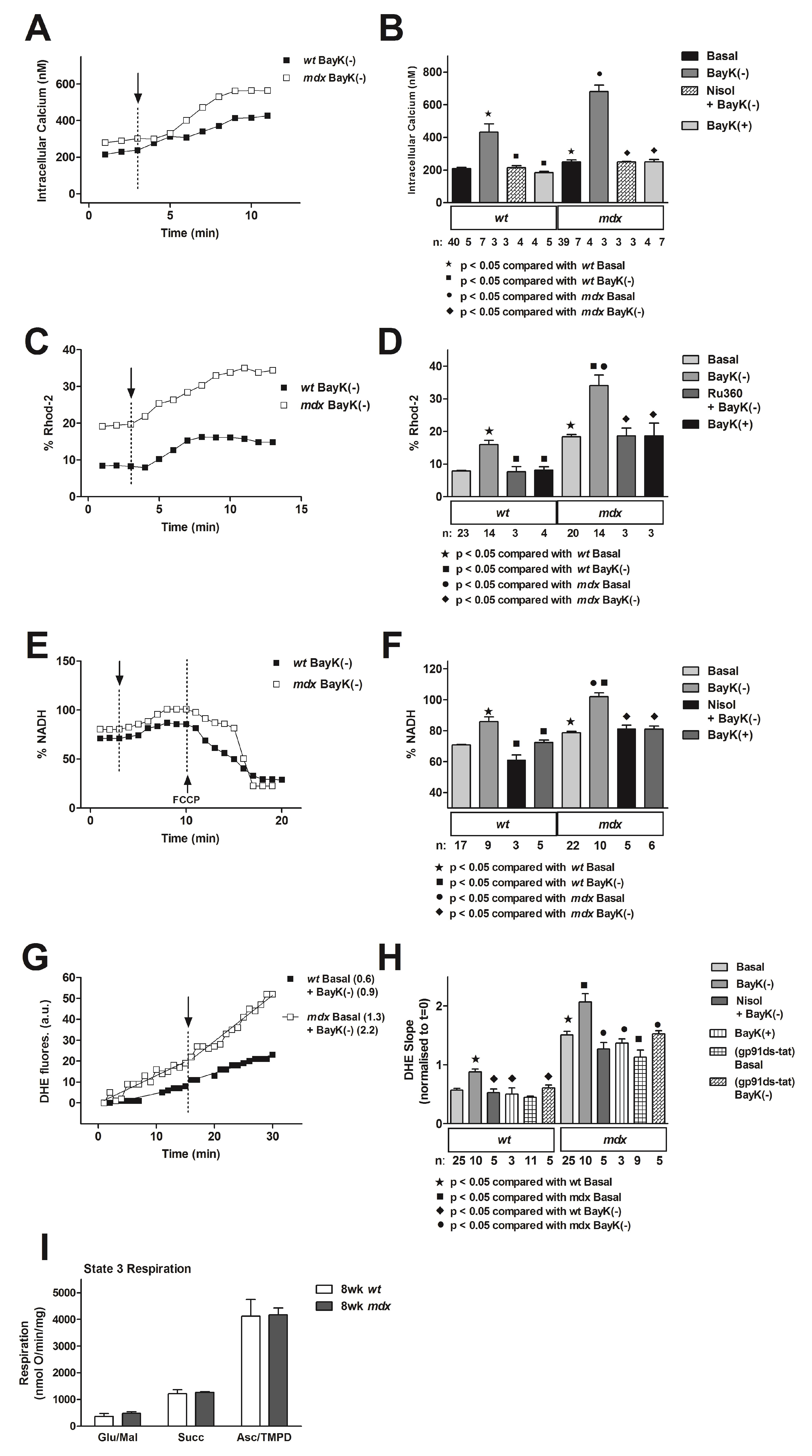
4.4. Altered Calcium Handling in the Mdx Mouse and Downstream Changes to Mitochondrial Function
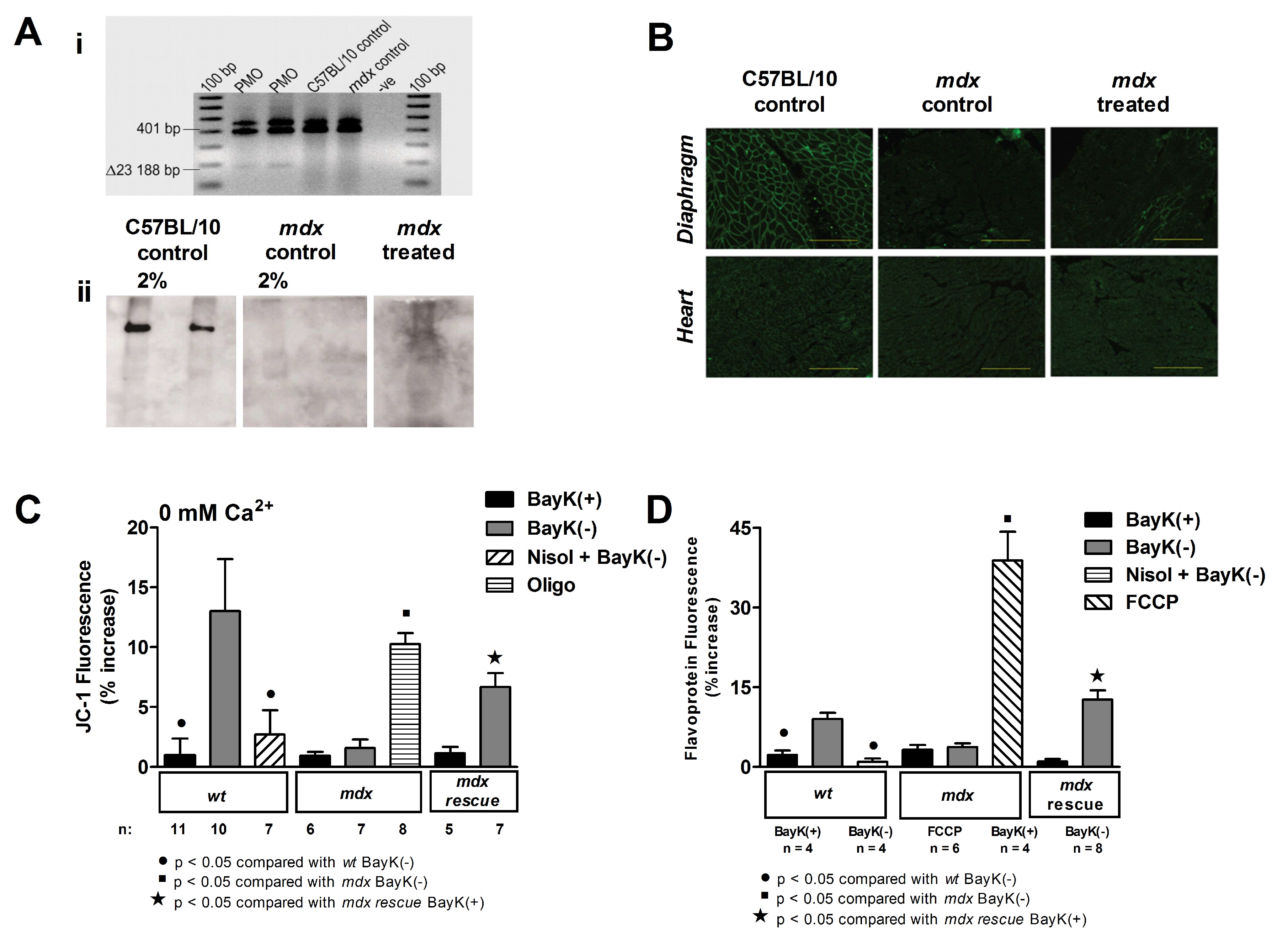
4.5. Recovery of Regulation of Mitochondrial Function by the L-Type Calcium Channel Following PMO Treatment
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). Prevalence of duchenne/becker muscular dystrophy among males aged 5–24 years—Four states, 2007. Morb. Mortal. Wkly. Rep. 2009, 58, 1119–1122. [Google Scholar]
- Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [PubMed]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of duchenne muscular dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Ararat, E.; Mhanna, M.J. Cardiac phenotype determines survival in duchenne muscular dystrophy. Pediatr. Pulmonol. 2016, 51, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Hor, K.N.; Taylor, M.D.; Al-Khalidi, H.R.; Cripe, L.H.; Raman, S.V.; Jefferies, J.L.; O’Donnell, R.; Benson, D.W.; Mazur, W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with duchenne muscular dystrophy: Effect of age and left ventricular systolic function. J. Cardiovasc. Magn. Reson. 2013, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Mazur, W.; Hor, K.N.; Germann, J.T.; Fleck, R.J.; Al-Khalidi, H.R.; Wansapura, J.P.; Chung, E.S.; Taylor, M.D.; Jefferies, J.L.; Benson, D.W.; et al. Patterns of left ventricular remodeling in patients with duchenne muscular dystrophy: A cardiac mri study of ventricular geometry, global function, and strain. Int. J. Cardiovasc. Imaging 2012, 28, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.C.; Pinto, Y.M.; Merkies, I.S.; de Die-Smulders, C.E.; Crijns, H.J.; Faber, C.G. Hereditary muscular dystrophies and the heart. Neuromusc. Disord. 2010, 20, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, A.; Miyagawa, M.; Yotsukura, M.; Tsuya, T.; Shirato, C.; Ishihara, T.; Aoyagi, T.; Ishikawa, K. The prevalence and prognostic significance of arrhythmias in duchenne type muscular dystrophy. Am. Heart J. 1992, 124, 1244–1250. [Google Scholar] [CrossRef]
- Chenard, A.A.; Becane, H.M.; Tertrain, F.; de Kermadec, J.M.; Weiss, Y.A. Ventricular arrhythmia in duchenne muscular dystrophy: Prevalence, significance and prognosis. Neuromusc. Disord. 1993, 3, 201–206. [Google Scholar] [CrossRef]
- Van Westering, T.L.; Betts, C.A.; Wood, M.J. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef] [PubMed]
- Amann, K.J.; Guo, A.W.; Ervasti, J.M. Utrophin lacks the rod domain actin binding activity of dystrophin. J. Biol. Chem. 1999, 274, 35375–35380. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Amann, K.J.; Renley, B.A.; Ervasti, J.M. A cluster of basic repeats in the dystrophin rod domain binds f-actin through an electrostatic interaction. J. Biol. Chem. 1998, 273, 28419–28423. [Google Scholar] [CrossRef] [PubMed]
- Duclos, F.; Straub, V.; Moore, S.A.; Venzke, D.P.; Hrstka, R.F.; Crosbie, R.H.; Durbeej, M.; Lebakken, C.S.; Ettinger, A.J.; van der Meulen, J.; et al. Progressive muscular dystrophy in α-sarcoglycan-deficient mice. J. Cell Biol. 1998, 142, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Rafael, J.A.; Chamberlain, J.S.; Campbell, K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997, 139, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Davies, S.M.; Filipovska, A.; Hool, L.C. L-type Ca(2+) channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H767–H775. [Google Scholar] [CrossRef] [PubMed]
- Pestronk, A.; Parhad, I.M.; Drachman, D.B.; Price, D.L. Membrane myopathy: Morphological similarities to duchenne muscular dystrophy. Muscle Nerve 1982, 5, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Mokri, B.; Engel, A.G. Duchenne dystrophy: Electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 1975, 25, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Bittner, R.E.; Leger, J.J.; Voit, T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol. 1992, 119, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Amthor, H.; Egelhof, T.; McKinnell, I.; Ladd, M.E.; Janssen, I.; Weber, J.; Sinn, H.; Schrenk, H.H.; Forsting, M.; Voit, T.; et al. Albumin targeting of damaged muscle fibres in the mdx mouse can be monitored by mri. Neuromusc. Disord. 2004, 14, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, C.; Loch, D.; Laws, N.; Trebbin, A.; Hoey, A.J. Timeline of cardiac dystrophy in 3–18-month-old mdx mice. Muscle Nerve 2010, 42, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Fanchaouy, M.; Polakova, E.; Jung, C.; Ogrodnik, J.; Shirokova, N.; Niggli, E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium 2009, 46, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P. Duchenne muscular dystrophy—What causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 2011, 43, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.L.; Williams, I.A.; Chu, Y.; Cooper, P.J.; Ju, Y.K.; Allen, D.G. Stretch-activated channels in the heart: Contributions to length-dependence and to cardiomyopathy. Prog. Biophys. Mol. Biol. 2008, 97, 232–249. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregon, A., Jr.; Lansman, J.B. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J. Physiol. 1994, 481, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Vandebrouck, C.; Duport, G.; Cognard, C.; Raymond, G. Cationic channels in normal and dystrophic human myotubes. Neuromusc. Disord. 2001, 11, 72–79. [Google Scholar] [CrossRef]
- Williams, I.A.; Allen, D.G. Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H846–H855. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Lee, D.I.; Hao, S.; Bedja, D.; Birnbaumer, L.; Cingolani, O.H.; Kass, D.A. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 2014, 114, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Lorin, C.; Vögeli, I.; Niggli, E. Dystrophic cardiomyopathy: Role of TRPV2 channels in stretch-induced cell damage. Cardiovasc. Res. 2015, 106, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.; Bode, E.; Venetucci, L.; Trafford, A. Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 2013, 58, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Lam, C.K.; Millay, D.P.; Sargent, M.A.; Hajjar, R.J.; Kranias, E.G.; Molkentin, J.D. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Investig. 2011, 121, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Morine, K.J.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Overexpression of serca1a in the mdx diaphragm reduces susceptibility to contraction-induced damage. Hum. Gene Ther. 2010, 21, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, S.; Zhang, X.; Li, J.; Ai, X.; Zhang, L.; Yu, D.; Ge, S.; Peng, Y.; Chen, X. Blunted cardiac beta-adrenergic response as an early indication of cardiac dysfunction in duchenne muscular dystrophy. Cardiovasc. Res. 2014, 103, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Rohman, M.S.; Emoto, N.; Takeshima, Y.; Yokoyama, M.; Matsuo, M. Decreased makap, ryanodine receptor, and SERCA2A gene expression in mdx hearts. Biochem. Biophys. Res. Commun. 2003, 310, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Niggli, E.; Ullrich, N.D.; Gutierrez, D.; Kyrychenko, S.; Polakova, E.; Shirokova, N. Posttranslational modifications of cardiac ryanodine receptors: Ca2+ signaling and ec-coupling. Biochim. Biophys. Acta 2013, 1833, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.; Lehnart, S.E.; Reiken, S.R.; Marks, A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004, 94, e61–e70. [Google Scholar] [CrossRef] [PubMed]
- Respress, J.L.; van Oort, R.J.; Li, N.; Rolim, N.; Dixit, S.S.; deAlmeida, A.; Voigt, N.; Lawrence, W.S.; Skapura, D.G.; Skardal, K.; et al. Role of RYR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 2012, 110, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Shan, J.; Betzenhauser, M.J.; Kushnir, A.; Reiken, S.; Meli, A.C.; Wronska, A.; Dura, M.; Chen, B.X.; Marks, A.R. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Investig. 2010, 120, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RYR2 trigger ventricular arrhythmias in duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Liberona, J.L.; Powell, J.A.; Shenoi, S.; Petherbridge, L.; Caviedes, R.; Jaimovich, E. Differences in both inositol 1,4,5-trisphosphate mass and inositol 1,4,5-trisphosphate receptors between normal and dystrophic skeletal muscle cell lines. Muscle Nerve 1998, 21, 902–909. [Google Scholar] [CrossRef]
- Mijares, A.; Altamirano, F.; Kolster, J.; Adams, J.A.; Lopez, J.R. Age-dependent changes in diastolic Ca2+ and Na+ concentrations in dystrophic cardiomyopathy: Role of Ca2+ entry and IP3. Biochem. Biophys. Res. Commun. 2014, 452, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Robert, V.; Massimino, M.L.; Tosello, V.; Marsault, R.; Cantini, M.; Sorrentino, V.; Pozzan, T. Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 2001, 276, 4647–4651. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.Q.; McNally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Engel, W.K. Muscle biopsies in neuromuscular diseases. Pediatr. Clin. N. Am. 1967, 14, 963–995. [Google Scholar] [CrossRef]
- Hathaway, P.W.; Engel, W.K.; Zellweger, H. Experimental myopathy after microarterial embolization: Comparison with childhood X-linked pseudohypertrophic muscular dystrophy. Arch. Neurol. 1970, 22, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Engel, W.K.; Derrer, E.C. Duchenne muscular dystrophy: Functional ischemia reproduces its characteristic lesions. Science 1971, 172, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Engel, W.K.; Derrer, E.C. Increased plasma enzyme concentrations in rats with functional ischaemia of muscle provide a possible model of duchenne muscular dystrophy. Nature 1972, 239, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, J.; Schneider, J.S.; Crassous, P.A.; Zheng, R.; Gonzalez, J.P.; Xie, L.H.; Beuve, A.; Fraidenraich, D.; Peluffo, R.D. Nitric oxide signalling pathway in duchenne muscular dystrophy mice: Up-regulation of L-arginine transporters. Biochem. J. 2013, 449, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Bia, B.L.; Cassidy, P.J.; Young, M.E.; Rafael, J.A.; Leighton, B.; Davies, K.E.; Radda, G.K.; Clarke, K. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 1999, 31, 1857–1862. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Michele, D.E. Dystrophin-glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of ampk signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 13663–13668. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. Alpha2 and alpha3 helices of dystrophin R16 and R17 frame a microdomain in the alpha1 helix of dystrophin R17 for neuronal NOS binding. Proc. Natl. Acad. Sci. USA 2013, 110, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Victor, R.G. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J. Physiol. 1998, 506, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Sander, M.; Lau, K.S.; Huang, P.L.; Stull, J.T.; Victor, R.G. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Shaul, P.W.; Yuhanna, I.S.; Froehner, S.C.; Adams, M.E. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ. Res. 2003, 92, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Chavoshan, B.; Harris, S.A.; Iannaccone, S.T.; Stull, J.T.; Thomas, G.D.; Victor, R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 13818–13823. [Google Scholar] [CrossRef] [PubMed]
- Chavoshan, B.; Sander, M.; Sybert, T.E.; Hansen, J.; Victor, R.G.; Thomas, G.D. Nitric oxide-dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J. Physiol. 2002, 540, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.A.; Barresi, R.; Byrne, B.J.; Tsimerinov, E.I.; Scott, B.L.; Walker, A.E.; Gurudevan, S.V.; Anene, F.; Elashoff, R.M.; Thomas, G.D.; et al. Tadalafil alleviates muscle ischemia in patients with becker muscular dystrophy. Sci. Transl. Med. 2012, 4, 162ra155. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Zhao, J.; Yue, Y.; Wasala, N.B.; Duan, D. Partial restoration of cardiac function with deltapdz nNOS in aged mdx model of duchenne cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D. Functional muscle ischemia in duchenne and becker muscular dystrophy. Front. Physiol. 2013, 4, 381. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Apoptotic pathways: The roads to ruin. Cell 1998, 94, 695–698. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Kroemer, G. Mitochondria in cell death: Novel targets for neuroprotection and cardioprotection. Trends Mol. Med. 2003, 9, 196–205. [Google Scholar] [CrossRef]
- Burelle, Y.; Khairallah, M.; Ascah, A.; Allen, B.G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Alterations in mitochondrial function as a harbinger of cardiomyopathy: Lessons from the dystrophic heart. J. Mol. Cell. Cardiol. 2010, 48, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.; Young, M.E.; Dyck, J.R.; Petrof, B.J.; Des Rosiers, C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Adams, A.M.; Davies, S.M.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Hool, L.C. Cross-talk between L-type Ca2+ channels and mitochondria. Clin. Exp. Pharmacol. Physiol. 2010, 37, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Hool, L.C. Role of the cytoskeleton in communication between L-type Ca2+ channels and mitochondria. Clin. Exp. Pharmacol. Physiol. 2013, 40, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, A.; Person, V.; Behlke, J.; Schaper, J.; Morano, I.; Haase, H. The carboxyl-terminal region of ahnak provides a link between cardiac l-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 2002, 16, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromusc. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Kieny, P.; Chollet, S.; Delalande, P.; Le Fort, M.; Magot, A.; Pereon, Y.; Perrouin Verbe, B. Evolution of life expectancy of patients with duchenne muscular dystrophy at afm yolaine de kepper centre between 1981 and 2011. Ann. Phys. Rehabil. Med. 2013, 56, 443–454. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Han, J.J.; Escolar, D.M.; Florence, J.M.; Duong, T.; Arrieta, A.; Clemens, P.R.; Hoffman, E.P.; et al. The cooperative international neuromuscular research group duchenne natural history study—A longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle Nerve 2013, 48, 32–54. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F. Cardiomyopathy of duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve 2011, 44, 8–19. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in duchenne muscular dystrophy. Working group of the national heart, lung, and blood institute in collaboration with parent project muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Muntoni, F.; Urtizberea, A.; Hughes, R.; Griggs, R. Report on the 124th enmc international workshop. Treatment of duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, The Netherlands. Neuromusc. Disord. 2004, 14, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Biggar, W.D.; Harris, V.A.; Eliasoph, L.; Alman, B. Long-term benefits of deflazacort treatment for boys with duchenne muscular dystrophy in their second decade. Neuromusc. Disord. 2006, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Moxley, R.T., 3rd; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical investigation of duchenne dystrophy group. Arch. Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Elia, M.; Carter, A.; Bacon, S.; Winearls, C.G.; Smith, R. Clinical usefulness of urinary 3-methylhistidine excretion in indicating muscle protein breakdown. Br. Med. J. 1981, 282, 351–354. [Google Scholar] [CrossRef]
- Rifai, Z.; Welle, S.; Moxley, R.T., 3rd; Lorenson, M.; Griggs, R.C. Effect of prednisone on protein metabolism in duchenne dystrophy. Am. J. Physiol. 1995, 268, E67–E74. [Google Scholar] [PubMed]
- Ball, E.H.; Sanwal, B.D. A synergistic effect of glucocorticoids and insulin on the differentiation of myoblasts. J. Cell. Physiol. 1980, 102, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.C.; Bootsma, A.L.; Willems, P.W.; Bar, P.R.; Wokke, J.H. Prednisone can protect against exercise-induced muscle damage. J. Neurol. 1996, 243, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.E.; Weber, M.; Vargas, C. Deflazacort increases laminin expression and myogenic repair, and induces early persistent functional gain in mdx mouse muscular dystrophy. Cell Transplant. 2000, 9, 551–564. [Google Scholar] [PubMed]
- Metzinger, L.; Passaquin, A.C.; Leijendekker, W.J.; Poindron, P.; Ruegg, U.T. Modulation by prednisolone of calcium handling in skeletal muscle cells. Br. J. Pharmacol. 1995, 116, 2811–2816. [Google Scholar] [CrossRef] [PubMed]
- Passaquin, A.C.; Lhote, P.; Ruegg, U.T. Calcium influx inhibition by steroids and analogs in c2c12 skeletal muscle cells. Br. J. Pharmacol. 1998, 124, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, F.; Guerin, C.; Blake, D.; Davies, K.; Karpati, G.; Holland, P. The effect of glucocorticoids on the accumulation of utrophin by cultured normal and dystrophic human skeletal muscle satellite cells. Neuromusc. Disord. 1995, 5, 105–114. [Google Scholar] [CrossRef]
- Barber, B.J.; Andrews, J.G.; Lu, Z.; West, N.A.; Meaney, F.J.; Price, E.T.; Gray, A.; Sheehan, D.W.; Pandya, S.; Yang, M.; et al. Oral corticosteroids and onset of cardiomyopathy in duchenne muscular dystrophy. J. Pediatr. 2013, 163, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for duchenne muscular dystrophy. Expert Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.H. Role of angiotensin receptor blockers in heart failure: Not yet resolvd. Circulation 1999, 100, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [PubMed]
- Delcayre, C.; Swynghedauw, B. Molecular mechanisms of myocardial remodeling. The role of aldosterone. J. Mol. Cell. Cardiol. 2002, 34, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Ishikawa, Y.; Ishikawa, Y.; Minami, R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in duchenne muscular dystrophy. J. Cardiol. 2009, 53, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Sechtem, U. Cardiac involvement in muscular dystrophy: Advances in diagnosis and therapy. Heart 2012, 98, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; River, L.P.; Pan, F.S.; Ji, L.; Wollmann, R.L. Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 4524–4528. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.; Turner, I.; Yasuda, S.; Martindale, J.; Davis, J.; Shillingford, M.; Kornegay, J.N.; Metzger, J.M. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Investig. 2010, 120, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.-J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Blake, D.J.; Roche, A.; Fairbrother, U.; Riss, J.; Byth, B.C.; Knight, A.E.; Kendrick-Jones, J.; Suthers, G.K.; Love, D.R.; et al. Primary structure of dystrophin-related protein. Nature 1992, 360, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Fairclough, R.J.; Bareja, A.; Davies, K.E. Progress in therapy for duchenne muscular dystrophy. Exp. Physiol. 2011, 96, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Moorwood, C.; Lozynska, O.; Suri, N.; Napper, A.D.; Diamond, S.L.; Khurana, T.S. Drug discovery for duchenne muscular dystrophy via utrophin promoter activation screening. PLoS ONE 2011, 6, e26169. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Bogdanovich, S.; Jensen, C.J.; Fischer, M.D.; Hansen-Schwartz, J.; Javazon, E.H.; Flake, A.W.; Edvinsson, L.; Khurana, T.S. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. USA 2004, 101, 13856–13860. [Google Scholar] [CrossRef] [PubMed]
- Voisin, V.; de la Porte, S. Pharmacological treatments for duchenne and becker dystrophies. J. Soc. Biol. 2005, 199, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Miura, P.; Chakkalakal, J.V.; Boudreault, L.; Belanger, G.; Hebert, R.L.; Renaud, J.M.; Jasmin, B.J. Pharmacological activation of pparbeta/delta stimulates utrophin a expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum. Mol. Genet. 2009, 18, 4640–4649. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, K.J.; Heun-Johnson, H.; Turner, A.J.; Baltgalvis, K.A.; Lowe, D.A.; Ervasti, J.M. Functional substitution by tat-utrophin in dystrophin-deficient mice. PLoS Med. 2009, 6, e1000083. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Rouviere, C.; Bonet-Kerrache, A. Rhoa leads to up-regulation and relocalization of utrophin in muscle fibers. Biochem. Biophys. Res. Commun. 2009, 384, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Amenta, A.R.; Yilmaz, A.; Bogdanovich, S.; McKechnie, B.A.; Abedi, M.; Khurana, T.S.; Fallon, J.R. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc. Natl. Acad. Sci. USA 2011, 108, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S. Read-through strategies for suppression of nonsense mutations in duchenne/becker muscular dystrophy: Aminoglycosides and ataluren (ptc124). J. Child Neurol. 2010, 25, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Swiderski, K.; Chamberlain, J.S. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve 2013, 47, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. Ptc124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hirawat, S.; Welch, E.M.; Elfring, G.L.; Northcutt, V.J.; Paushkin, S.; Hwang, S.; Leonard, E.M.; Almstead, N.G.; Ju, W.; Peltz, S.W.; et al. Safety, tolerability, and pharmacokinetics of ptc124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J. Clin. Pharmacol. 2007, 47, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Li, S.; Harper, S.Q.; Welikson, R.; Bourque, D.; DelloRusso, C.; Hauschka, S.D.; Chamberlain, J.S. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromusc. Disord. 2002, 12, S23–S29. [Google Scholar] [CrossRef]
- Yue, Y.; Li, Z.; Harper, S.Q.; Davisson, R.L.; Chamberlain, J.S.; Duan, D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation 2003, 108, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, S.; Bauer, R.; Bekeredjian, R.; Stucka, R.; Rutschow, D.; Lochmuller, H.; Kleinschmidt, J.A.; Katus, H.A.; Muller, O.J. Long-term preservation of cardiac structure and function after adeno-associated virus serotype 9-mediated microdystrophin gene transfer in mdx mice. Hum. Gene Ther. 2012, 23, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by raav9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.; Blankinship, M.J.; Allen, J.M.; Gregorevic, P.; Chamberlain, J.S.; Metzger, J.M. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Shin, J.H.; Yue, Y.; Wasala, N.B.; Lai, Y.; Duan, D. Aav micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of duchenne muscular dystrophy cardiomyopathy. J. Mol. Cell. Cardiol. 2012, 53, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Shin, J.H.; Yue, Y.; Duan, D. Aav-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.; Sahenk, Z.; Malik, V.; Kaspar, B.K.; Walker, C.M.; Clark, K.R. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosci. Lett. 2012, 527, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne′s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Popplewell, L.; Dickson, G. Genetic therapeutic approaches for duchenne muscular dystrophy. Hum. Gene Ther. 2012, 23, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, A.; Wells, K.E.; Wells, D.J. Immune responses to dystropin: Implications for gene therapy of duchenne muscular dystrophy. Gene Ther. 2000, 7, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Lorain, S.; Gross, D.A.; Goyenvalle, A.; Danos, O.; Davoust, J.; Garcia, L. Transient immunomodulation allows repeated injections of aav1 and correction of muscular dystrophy in multiple muscles. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Kissel, J.T.; Amato, A.A.; King, W.; Signore, L.; Prior, T.W.; Sahenk, Z.; Benson, S.; McAndrew, P.E.; Rice, R.; et al. Myoblast transfer in the treatment of duchenne’s muscular dystrophy. N. Engl. J. Med. 1995, 333, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Dell’Agnola, C.; Wang, Z.; Storb, R.; Tapscott, S.J.; Kuhr, C.S.; Hauschka, S.D.; Lee, R.S.; Sale, G.E.; Zellmer, E.; Gisburne, S.; et al. Hematopoietic stem cell transplantation does not restore dystrophin expression in duchenne muscular dystrophy dogs. Blood 2004, 104, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered dmd patient stem cells into dystrophic mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’Antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007, 16, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Huard, C.; Moisset, P.A.; Dicaire, A.; Merly, F.; Tardif, F.; Asselin, I.; Tremblay, J.P. Transplantation of dermal fibroblasts expressing myod1 in mouse muscles. Biochem. Biophys. Res. Commun. 1998, 248, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Razini, P.; Erratico, S.; Torrente, Y.; Meregalli, M. Cell based therapy for duchenne muscular dystrophy. J. Cell. Physiol. 2009, 221, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Huard, J.; Bouchard, J.P.; Roy, R.; Malouin, F.; Dansereau, G.; Labrecque, C.; Albert, N.; Richards, C.L.; Lemieux, B.; Tremblay, J.P. Human myoblast transplantation: Preliminary results of 4 cases. Muscle Nerve 1992, 15, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, J.; Chapdelaine, P.; Boisvert, S.; Almeida, L.P.; Corbeil, J.; Montpetit, A.; Tremblay, J.P. Endonucleases: Tools to correct the dystrophin gene. J. Gene Med. 2011, 13, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by talen and crispr-cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Popplewell, L.; Koo, T.; Leclerc, X.; Duclert, A.; Mamchaoui, K.; Gouble, A.; Mouly, V.; Voit, T.; Paques, F.; Cedrone, F.; et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum. Gene Ther. 2013, 24, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R. Duchenne muscular dystrophy: Crispr/cas9 treatment. Cell Res. 2016, 26, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sane, H.; Paranjape, A.; Bhagawanani, K.; Gokulchandran, N.; Badhe, P. Autologous bone marrow mononuclear cell transplantation in duchenne muscular dystrophy—A case report. Am. J. Case Rep. 2014, 15, 128–134. [Google Scholar] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Piette, V.; Côté, C.H.; Chapdelaine, P.; Hogrel, J.-Y.; Paradis, M.; Bouchard, J.-P.; Sylvain, M.; et al. First test of a “high-density injection” protocol for myogenic cell transplantation throughout large volumes of muscles in a duchenne muscular dystrophy patient: Eighteen months follow-up. Neuromusc. Disord. 2007, 17, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Reutenauer-Patte, J.; Boittin, F.X.; Patthey-Vuadens, O.; Ruegg, U.T.; Dorchies, O.M. Urocortins improve dystrophic skeletal muscle structure and function through both pka- and epac-dependent pathways. Am. J. Pathol. 2012, 180, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Amthor, H.; Hoogaars, W.M. Interference with myostatin/actriib signaling as a therapeutic strategy for duchenne muscular dystrophy. Curr. Gene Ther. 2012, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Mourkioti, F.; Kustan, J.; Kraft, P.; Day, J.W.; Zhao, M.-M.; Kost-Alimova, M.; Protopopov, A.; DePinho, R.A.; Bernstein, D.; Meeker, A.K.; et al. Role of telomere dysfunction in cardiac failure in duchenne muscular dystrophy. Nat. Cell Biol. 2013, 15, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. 2008, 586, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Delfín, D.A.; Xu, Y.; Peterson, J.M.; Guttridge, D.C.; Rafael-Fortney, J.A.; Janssen, P.M. Improvement of cardiac contractile function by peptide-based inhibition of NF-kB in the utrophin/dystrophin-deficient murine model of muscular dystrophy. J. Transl. Med. 2011, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, C.; Hollinger, K.; Selsby, J.T.; Amin, R.; Quindry, J.C. Histological and biochemical outcomes of cardiac pathology in mdx mice with dietary quercetin enrichment. Exp. Physiol. 2015, 100, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.L.; Swiderski, K.; Murphy, K.T.; Gehrig, S.M.; Curl, C.L.; Chandramouli, C.; Febbraio, M.A.; Delbridge, L.M.; Koopman, R.; Lynch, G.S. Bgp-15 improves aspects of the dystrophic pathology in mdx and dko mice with differing efficacies in heart and skeletal muscle. Am. J. Pathol. 2016, 186, 3246–3260. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Veedu, R.N.; Fletcher, S. The emperor’s new dystrophin: Finding sense in the noise. Trends Mol. Med. 2015, 21, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon skipping for nonsense mutations in duchenne muscular dystrophy: Too many mutations, too few patients? Expert Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. Rna targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Honeyman, K.; Cheng, A.J.; Ly, T.; Lloyd, F.; Fletcher, S.; Morgan, J.E.; Partridge, T.A.; Wilton, S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 2002, 4, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of pro051 in duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of duchenne muscular dystrophy (demand ii): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- GlaxoSmithKline. A Clinical Study to Assess the Efficacy and Safety of GSK2402968 in Subjects with Duchenne Muscular Dystrophy; U.S. National Institutes of Health: Bethesda, MD, USA, 2013.
- Flanigan, K.M.; Voit, T.; Rosales, X.Q.; Servais, L.; Kraus, J.E.; Wardell, C.; Morgan, A.; Dorricott, S.; Nakielny, J.; Quarcoo, N.; et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with duchenne muscular dystrophy: Results of a double-blind randomized clinical trial. Neuromusc. Disord. 2014, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Kim, D.S.; Kwon, H.J. CG sequence- and phosphorothioate backbone modification-dependent activation of the nf-kappab-responsive gene expression by cpg-oligodeoxynucleotides in human rpmi 8226 b cells. Mol. Immunol. 2004, 41, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Alvarez, R.; Roderiquez, G.; Guan, E.; Caldwell, Q.; Wang, J.; Phelan, M.; Norcross, M.A. Cpg-independent synergistic induction of beta-chemokines and a dendritic cell phenotype by orthophosphorothioate oligodeoxynucleotides and granulocyte-macrophage colony-stimulating factor in elutriated human primary monocytes. J. Immunol. 2005, 174, 6113–6121. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.S.; Sobry, C.; Derr, V.; Adams, M.J.; Besten, C.D.; De Kimpe, S.; Francis, I.; Gales, T.L.; Haworth, R.; Maguire, S.R.; et al. Species-specific inflammatory responses as a primary component for the development of glomerular lesions in mice and monkeys following chronic administration of a second-generation antisense oligonucleotide. Toxicol. Pathol. 2014, 42, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krieg, A.M. Exon skipping therapy for duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 2015, 87, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Iversen, P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005, 5, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Gebski, B.L.; Mann, C.J.; Fletcher, S.; Wilton, S.D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 2003, 12, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lu, P.; Benrashid, E.; Malik, S.; Ashar, J.; Doran, T.J.; Lu, Q.L. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2010, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Sharp, P.S.; Graham, I.R.; Arechavala-Gomeza, V.; Foster, K.; Muntoni, F.; Wells, D.J.; Dickson, G. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer avi-4658 in duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Pane, M.; Mazzone, E.S.; Sormani, M.P.; Messina, S.; Vita, G.L.; Fanelli, L.; Berardinelli, A.; Torrente, Y.; D’Amico, A.; Lanzillotta, V.; et al. 6 minute walk test in duchenne md patients with different mutations: 12 month changes. PLoS ONE 2014, 9, e83400. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in duchenne muscular dystrophy: Pathogenesis and therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Margus, H.; Padari, K.; Pooga, M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercised induced cardiomyopathy following pip-pmo treatment in dystrophic mdx mice. Sci. Rep. 2015, 5, 8986. [Google Scholar] [CrossRef] [PubMed]
- Wasala, N.B.; Yue, Y.; Vance, J.; Duan, D. Uniform low-level dystrophin expression in the heart partially preserved cardiac function in an aged mouse model of duchenne cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 102, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Meadows, L.S.; Isom, L.L. Sodium channels as macromolecular complexes: Implications for inherited arrhythmia syndromes. Cardiovasc. Res. 2005, 67, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Jordan, M.C.; Roos, K.P.; Hool, L.C. Decreased myocardial injury and improved contractility after administration of a peptide derived against the alpha-interacting domain of the L-type calcium channel. J. Am. Heart Assoc. 2014, 3, e000961. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Hool, L.C. How does calcium regulate mitochondrial energetics in the heart?—New insights. Heart Lung Circ. 2014, 23, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.; Johnstone, V.; Szappanos, H.C.; Richman, T.; Tsoutsman, T.; Filipovska, A.; Semsarian, C.; Hool, L. The L-type Ca2+ channel facilitates abnormal metabolic activity in the cTni-G203S mouse model of hypertrophic cardiomyopathy. J. Physiol. 2016, 594, 4051–4070. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Lader, A.S.; Kwiatkowski, D.J.; Cantiello, H.F. Role of gelsolin in the actin filament regulation of cardiac L-type calcium channels. Am. J. Physiol. 1999, 277, C1277–C1283. [Google Scholar] [PubMed]
- Rueckschloss, U.; Isenberg, G. Cytochalasin d reduces Ca2+ currents via cofilin-activated depolymerization of F-actin in guinea-pig cardiomyocytes. J. Physiol. 2001, 537, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, L.; Oliviero, P.; Samuel, J.L. Cytoskeleton and mitochondrial morphology and function. Mol. Cell. Biochem. 1998, 184, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc. Med. 2002, 12, 339–348. [Google Scholar] [CrossRef]
- Maloyan, A.; Sanbe, A.; Osinska, H.; Westfall, M.; Robinson, D.; Imahashi, K.; Murphy, E.; Robbins, J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-b-crystallin desmin-related cardiomyopathy. Circulation 2005, 112, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Hardy, N.; Viola, H.M.; Johnstone, V.P.; Clemons, T.D.; Cserne Szappanos, H.; Singh, R.; Smith, N.M.; Iyer, K.S.; Hool, L.C. Nanoparticle-mediated dual delivery of an antioxidant and a peptide against the L-type Ca2+ channel enables simultaneous reduction of cardiac ischemia-reperfusion injury. ACS Nano 2015, 9, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Tsoutsman, T.; Chung, J.; Doolan, A.; Nguyen, L.; Williams, I.A.; Tu, E.; Lam, L.; Bailey, C.G.; Rasko, J.E.; Allen, D.G.; et al. Molecular insights from a novel cardiac troponin i mouse model of familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2006, 41, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Johnstone, V.P.A.; Cserne Szappanos, H.; Richman, T.R.; Tsoutsman, T.; Filipovska, A.; Semsarian, C.; Seidman, J.G.; Seidman, C.E.; Hool, L.C. The role of the L-type Ca2+ channel in altered metabolic activity in a murine model of hypertrophic cardiomyopathy. JACC Basic Transl. Sci. 2016, 1, 61–72. [Google Scholar] [CrossRef]
- Phillips, M.F.; Quinlivan, R. Calcium antagonists for duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Koenig, X.; Dysek, S.; Kimbacher, S.; Mike, A.K.; Cervenka, R.; Lukacs, P.; Nagl, K.; Dang, X.B.; Todt, H.; Bittner, R.E.; et al. Voltage-gated ion channel dysfunction precedes cardiomyopathy development in the dystrophic heart. PLoS ONE 2011, 6, e20300. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Rubi, L.; Obermair, G.J.; Cervenka, R.; Dang, X.B.; Lukacs, P.; Kummer, S.; Bittner, R.E.; Kubista, H.; Todt, H.; et al. Enhanced currents through L-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H564–H573. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Matsuzaki, T.; Date, M.; Wada, K. Skeletal muscle fiber degeneration in mdx mice induced by electrical stimulation. Muscle Nerve 1997, 20, 1422–1432. [Google Scholar] [CrossRef]
- Sadeghi, A.; Doyle, A.D.; Johnson, B.D. Regulation of the cardiac L-type Ca2+ channel by the actin-binding proteins alpha-actinin and dystrophin. Am. J. Physiol. Cell Physiol 2002, 282, C1502–C1511. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Byerly, L. Calcium channel. Annu. Rev. Neurosci. 1981, 4, 69–125. [Google Scholar] [CrossRef] [PubMed]
- Woolf, P.J.; Lu, S.; Cornford-Nairn, R.; Watson, M.; Xiao, X.H.; Holroyd, S.M.; Brown, L.; Hoey, A.J. Alterations in dihydropyridine receptors in dystrophin-deficient cardiac muscle. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2439–H2445. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Andersson, D.C.; Lanner, J.T. Can’t live with or without it: Calcium and its role in duchenne muscular dystrophy-induced muscle weakness. Focus on “serca1 overexpression minimizes skeletal muscle damage in dystrophic mouse models”. Am. J. Physiol. Cell Physiol. 2015, 308, C697–C698. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnstone, V.P.A.; Viola, H.M.; Hool, L.C. Dystrophic Cardiomyopathy—Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies. Genes 2017, 8, 108. https://doi.org/10.3390/genes8040108
Johnstone VPA, Viola HM, Hool LC. Dystrophic Cardiomyopathy—Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies. Genes. 2017; 8(4):108. https://doi.org/10.3390/genes8040108
Chicago/Turabian StyleJohnstone, Victoria P. A., Helena M. Viola, and Livia C. Hool. 2017. "Dystrophic Cardiomyopathy—Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies" Genes 8, no. 4: 108. https://doi.org/10.3390/genes8040108
APA StyleJohnstone, V. P. A., Viola, H. M., & Hool, L. C. (2017). Dystrophic Cardiomyopathy—Potential Role of Calcium in Pathogenesis, Treatment and Novel Therapies. Genes, 8(4), 108. https://doi.org/10.3390/genes8040108