
MYC in Regulating Immunity: Metabolism and Beyond

{kind=link}
{kind=link}
Abstract
:1. Introduction
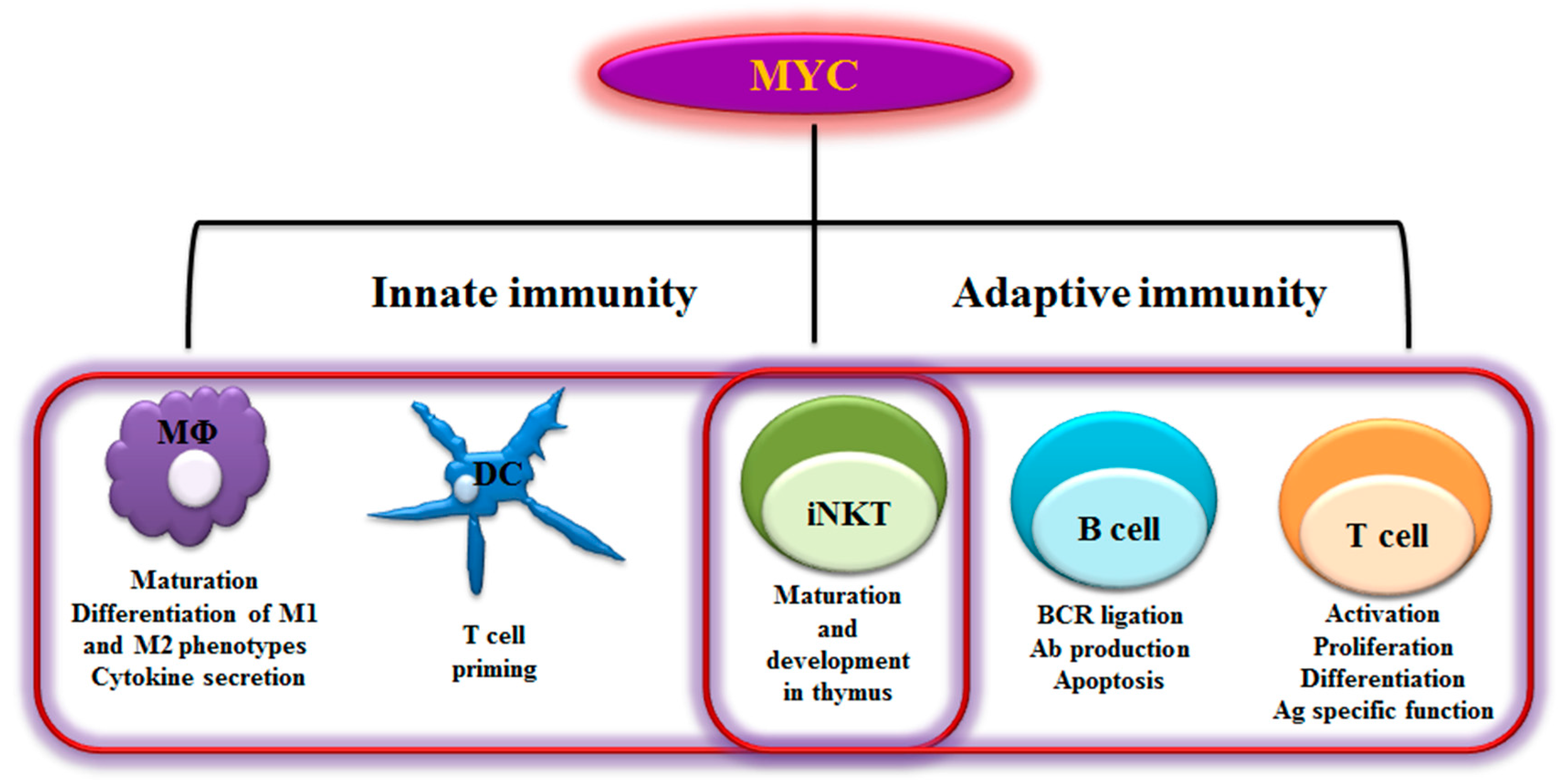
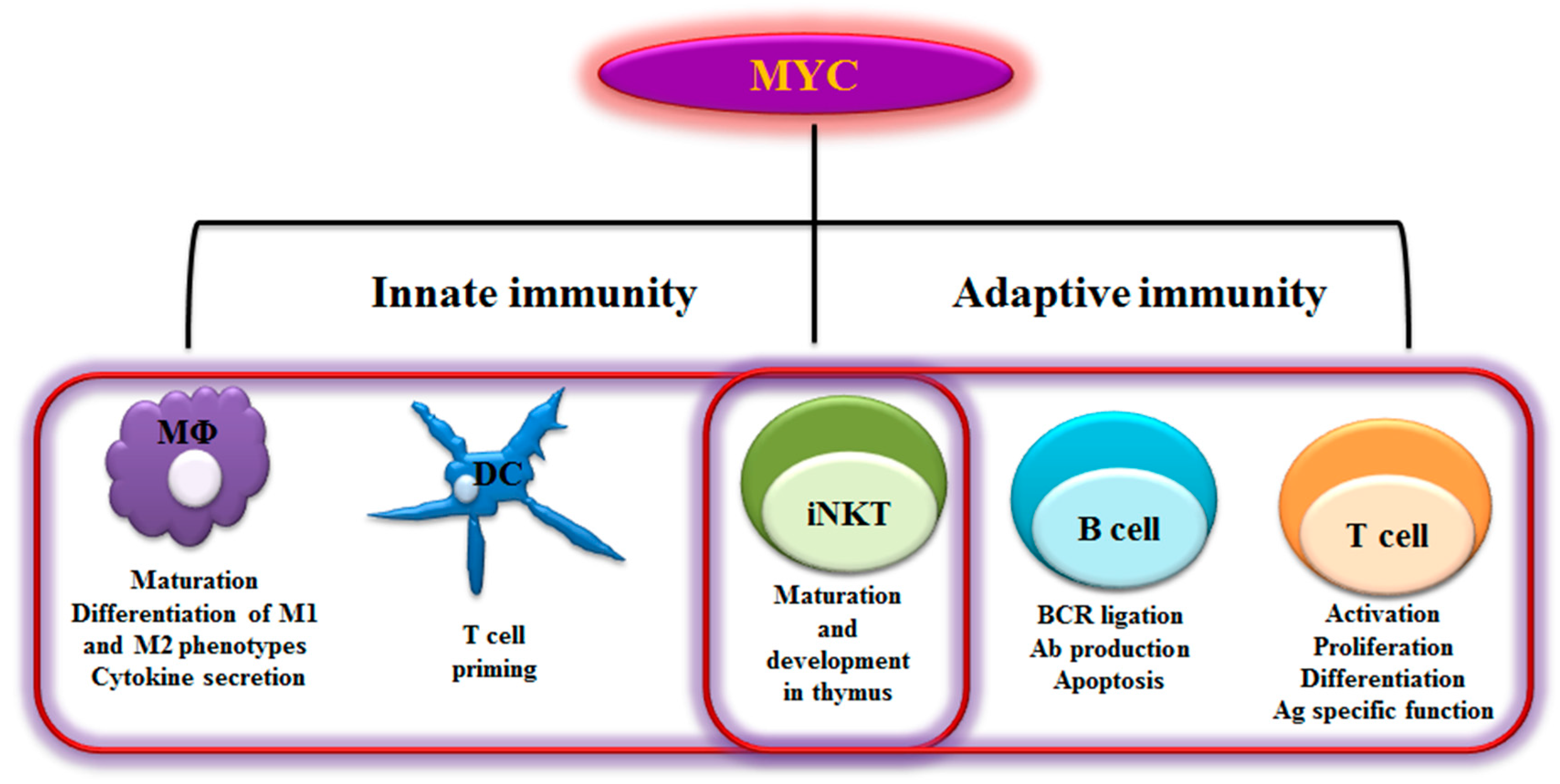
2. MYC in Immunity
2.1 B Lymphocytes
2.2 T Lymphocytes
2.3 NKT Cells
2.4 Dendritic Cells
2.5 Macrophages
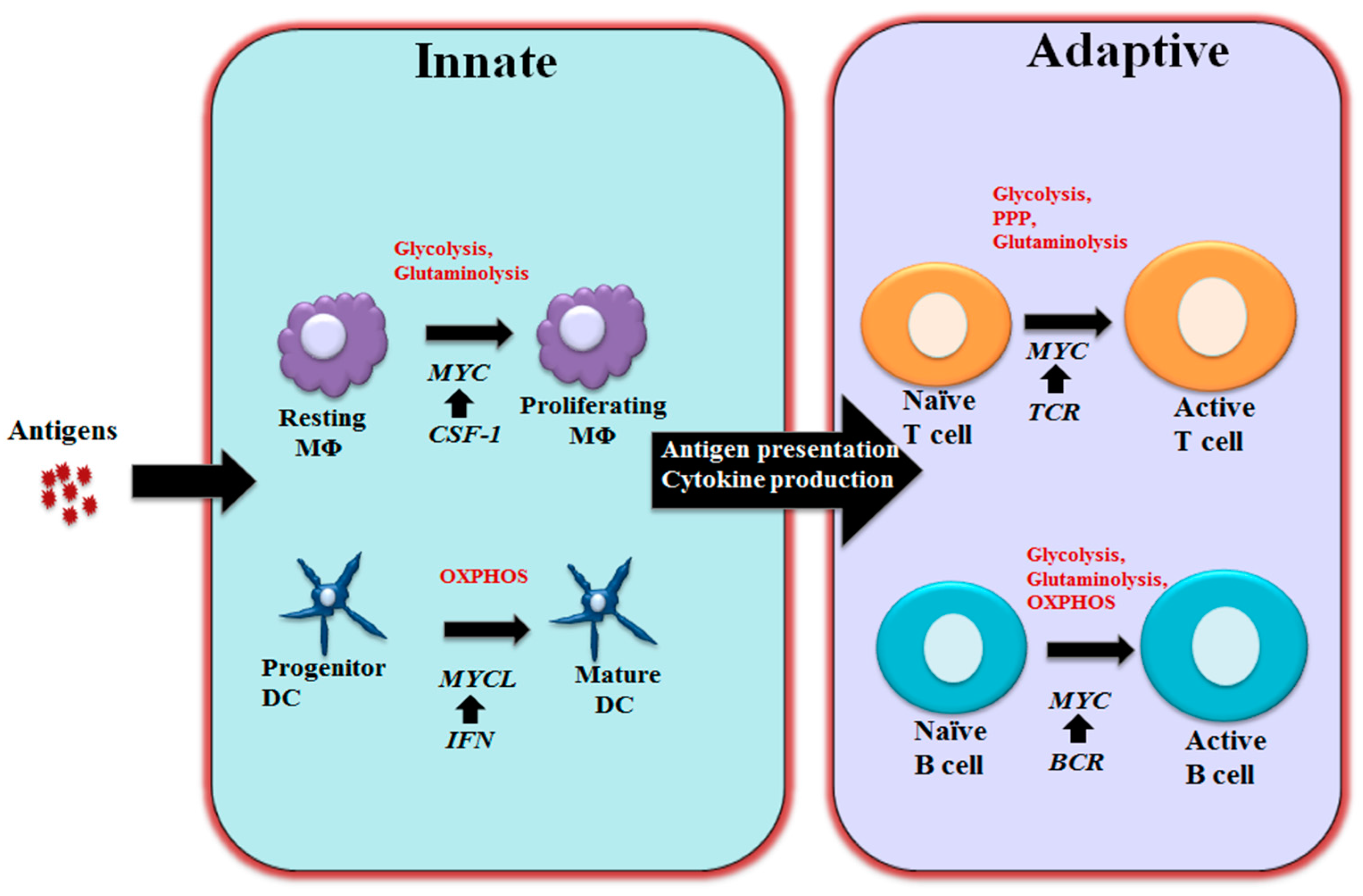
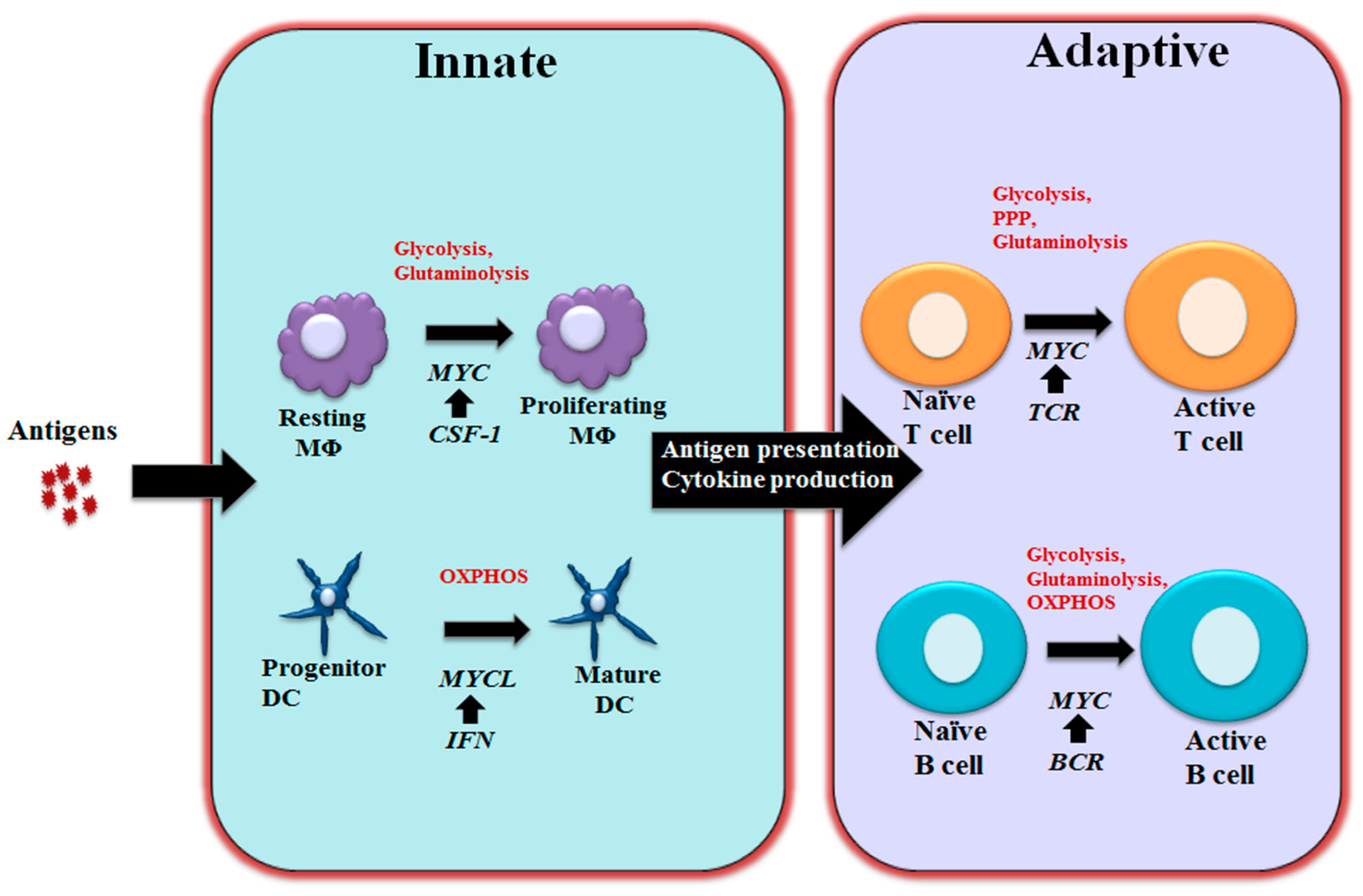
3. MYC in Regulating Immune Metabolism
3.1 MYC in Regulating T and B Cell Metabolism
3.2 MYC in Regulating Macrophage and Dendritic Cells Metabolism
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hayward, W.S.; Neel, B.G.; Astrin, S.M. Activation of a cellular onc gene by promoter insertion in alv-induced lymphoid leukosis. Nature 1981, 290, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [PubMed]
- Dang, C.V. C-myc target genes involved in cell growth, apoptosis, and metabolism. J. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Dang, C.V. Myc on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Dangm, C.V.; O’Donell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-myc target gene network. Seminar Cancer Biol. 2006, 16, 253–264. [Google Scholar]
- Dang, C.V.; Resar, L.M.; Emison, E.; Kim, S.; Li, Q.; Prescott, J.E.; Wonsey, D.; Zeller, K. Function of the c-myc oncogenic transcription factor. Exp. Cell. Res. 1999, 253, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. C-myc: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. Myc oncogenes and human neoplastic disease. Oncogene 1999, 18. [Google Scholar] [CrossRef] [PubMed]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of myc and its interactome. Csh. Perspect. Med. 2014, 4, a014357. [Google Scholar]
- Ladanyi, M.; Park, C.K.; Lewis, R.; Jhanwar, S.C.; Healey, J.H.; Huvos, A.G. Sporadic amplification of the myc gene in human osteosarcomas. Diagn. Mol. Pathol. 1993, 2, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 1991, 56, 1–48. [Google Scholar] [PubMed]
- Escot, C.; Theillet, C.; Lidereau, R.; Spyratos, F.; Champeme, M.-H.; Gest, J.; Callahan, R. Genetic alteration of the c-myc protooncogene (myc) in human primary breast carcinomas. Proc. Natl. Acad. Sci. 1986, 83, 4834–4838. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Ellison, J.; Busch, M.; Rosenau, W.; Varmus, H.E.; Bishop, J.M. Enhanced expression of the human gene n-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc. Natl. Acad. Sci. 1984, 81, 4940–4944. [Google Scholar] [CrossRef] [PubMed]
- Schlagbauer-Wadl, H.; Griffioen, M.; van Elsas, A.; Schrier, P.I.; Pustelnik, T.; Eichler, H.-G.; Wolff, K.; Pehamberger, H.; Jansen, B. Influence of increased c-myc expression on the growth characteristics of human melanoma. J. Invest. Dermatol. 1999, 112, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The myc/max/mad network and the transcriptional control of cell behavior. Annu. Rev. Cell. Dev. Bi. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-myc protein requires dimerization with max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Amati, B.; Littlewood, T.; Evan, G.; Land, H. The c-myc protein induces cell cycle progression and apoptosis through dimerization with max. EMBO J. 1993, 12, 5083. [Google Scholar] [PubMed]
- Foley, K.P.; Eisenman, R.N. Two mad tails: What the recent knockouts of mad1 and mxi1 tell us about the myc/max/mad network. Biochim. Biophys. Acta, Rev. Cancer 1999, 1423, M37–M47. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Parkin, J.; Cohen, B. An overview of the immune system. The Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Niiro, H.; Clark, E.A. Regulation of b-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002, 2, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A. Antigen-specific interaction between t and b cells. Nature 1984, 314, 537–539. [Google Scholar] [CrossRef]
- Murn, J.; Mlinaric-Rascan, I.; Vaigot, P.; Alibert, O.; Frouin, V.; Gidrol, X. A myc-regulated transcriptional network controls b-cell fate in response to bcr triggering. BMC genomics 2009, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.J.; Strasser, A.; Gerondakis, S. B cell growth is controlled by phosphatidylinosotol 3-kinase-dependent induction of rel/nf-κb regulated c-myc transcription. Mol. Cell 2002, 10, 1283–1294. [Google Scholar] [CrossRef]
- Grumont, R.; Lock, P.; Mollinari, M.; Shannon, F.M.; Moore, A.; Gerondakis, S. The mitogen-induced increase in t cell size involves pkc and nfat activation of rel/nf-κb-dependent c-myc expression. Immunity 2004, 21, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Habib, T.; Park, H.; Tsang, M.; de Alborán, I.M.; Nicks, A.; Wilson, L.; Knoepfler, P.S.; Andrews, S.; Rawlings, D.J.; Eisenman, R.N. Myc stimulates b lymphocyte differentiation and amplifies calcium signaling. J. Cell Biol. 2007, 179, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Iritani, B.M.; Eisenman, R.N. C-myc enhances protein synthesis and cell size during b lymphocyte development. Proc. Natl. Acad. Sci. 1999, 96, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- de Alboran, I.M.; O'Hagan, R.C.; Gärtner, F.; Malynn, B.; Davidson, L.; Rickert, R.; Rajewsky, K.; DePinho, R.A.; Alt, F.W. Analysis of c-myc function in normal cells via conditional gene-targeted mutation. Immunity 2001, 14, 45–55. [Google Scholar] [CrossRef]
- de Alborán, I.M.; Baena, E.; Martinez-A, C. C-myc-deficient b lymphocytes are resistant to spontaneous and induced cell death. Cell Death Differ. 2004, 11, 61–68. [Google Scholar] [CrossRef] [PubMed]
- von Andrian, U.H.; Mackay, C.R. T-cell function and migration—two sides of the same coin. N. Engl. J. Med. 2000, 343, 1020–1034. [Google Scholar] [PubMed]
- Hall, J.; Morris, B. The immediate effect of antigens on the cell output of a lymph node. Br. J. Exp. Pathol. 1965, 46, 450. [Google Scholar] [PubMed]
- Itano, A.A.; Jenkins, M.K. Antigen presentation to naive cd4 t cells in the lymph node. Nat. Immunol. 2003, 4, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.C.; Sinclair, L.V.; Kaskar, A.; Hukelmann, J.L.; Navarro, M.N.; Ferrero, I.; MacDonald, H.R.; Cowling, V.H.; Cantrell, D.A. Single cell tuning of myc expression by antigen receptor signal strength and interleukin-2 in t lymphocytes. EMBO J. 2015, e201490252. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; June, C.H.; Thompson, C.B. Multiple mechanisms regulate c-myc gene expression during normal t cell activation. EMBO J. 1988, 7, 2787. [Google Scholar] [PubMed]
- Iritani, B.M.; Delrow, J.; Grandori, C.; Gomez, I.; Klacking, M.; Carlos, L.S.; Eisenman, R.N. Modulation of t-lymphocyte development, growth and cell size by the myc antagonist and transcriptional repressor mad1. EMBO J. 2002, 21, 4820–4830. [Google Scholar] [CrossRef] [PubMed]
- Dose, M.; Khan, I.; Guo, Z.; Kovalovsky, D.; Krueger, A.; von Boehmer, H.; Khazaie, K.; Gounari, F. C-myc mediates pre-tcr-induced proliferation but not developmental progression. Blood 2006, 108, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C. A notch1-driven myc enhancer promotes t cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- La Starza, R.; Borga, C.; Barba, G.; Pierini, V.; Schwab, C.; Matteucci, C.; Fernandez, A.G.L.; Leszl, A.; Cazzaniga, G.; Chiaretti, S. Genetic profile of t-cell acute lymphoblastic leukemias with myc-translocations. Blood 2014. blood-2014–06–578856. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by myc in hematopoietic lineages. Mol Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Jiang, W.; Ferrero, I.; Laurenti, E.; Trumpp, A.; MacDonald, H.R. C-myc controls the development of cd8αα tcrαβ intestinal intraepithelial lymphocytes from thymic precursors by regulating il-15–dependent survival. Blood 2010, 115, 4431–4438. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R. C-myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Pinto, A.K.; Curtis, J.D.; Persaud, S.P.; Cella, M.; Lin, C.-C.; Edelson, B.T.; Allen, P.M.; Colonna, M.; Pearce, E.L. C-myc-induced transcription factor ap4 is required for host protection mediated by cd8+ t cells. Nat. Immunol. 2014, 15, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Pellicci, D.G.; Hammond, K.J.; Uldrich, A.P.; Baxter, A.G.; Smyth, M.J.; Godfrey, D.I. A natural killer t (nkt) cell developmental pathway involving a thymus-dependent nk1. 1− cd4+ cd1d-dependent precursor stage. J. Exp. Med. 2002, 195, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of nkt cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Rivera, M.N.; Park, S.-H.; Roark, J.H. Mouse cd1-specific nk1 t cells: Development, specificity, and function. Annu Rev. Immunol 1997, 15, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Dose, M.; Sleckman, B.P.; Han, J.; Bredemeyer, A.L.; Bendelac, A.; Gounari, F. Intrathymic proliferation wave essential for vα14+ natural killer t cell development depends on c-myc. Proc. Natl. Acad. Sci. 2009, 106, 8641–8646. [Google Scholar] [CrossRef] [PubMed]
- Savage, A.K.; Constantinides, M.G.; Han, J.; Picard, D.; Martin, E.; Li, B.; Lantz, O.; Bendelac, A. The transcription factor plzf directs the effector program of the nkt cell lineage. Immunity 2008, 29, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Mycko, M.P.; Ferrero, I.; Wilson, A.; Jiang, W.; Bianchi, T.; Trumpp, A.; MacDonald, H.R. Selective requirement for c-myc at an early stage of vα14i nkt cell development. J. Immunol. 2009, 182, 4641–4648. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu Rev. Immunol 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Nussenzweig, M.C. Origin and development of dendritic cells. Immunol Rev. 2010, 234, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wumesh, K.; Satpathy, A.T.; Rapaport, A.S.; Briseño, C.G.; Wu, X.; Albring, J.C.; Russler-Germain, E.V.; Kretzer, N.M.; Durai, V.; Persaud, S.P. L-myc expression by dendritic cells is required for optimal t-cell priming. Nature 2014, 507, 243–247. [Google Scholar]
- Becker, A.M.; Michael, D.G.; Satpathy, A.T.; Sciammas, R.; Singh, H.; Bhattacharya, D. Irf-8 extinguishes neutrophil production and promotes dendritic cell lineage commitment in both myeloid and lymphoid mouse progenitors. Blood 2012, 119, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Pluddemann, A. Tissue macrophage heterogeneity: Issues and prospects. Semin Immunopathol 2013, 35, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu Rev. Immunol 2009, 27, 669–692. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu Rev. Immunol 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from myc-dependent to hif1α-dependent. Proc. Natl. Acad. Sci. 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.F.; Cleveland, J.L.; Shurtleff, S.A.; Sherr, C.J. Myc rescue of a mutant csf-1 receptor impaired in mitogenic signalling. Nature 1991, 353, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. Csf-1 and cell cycle control in macrophages. Mol Reprod. Dev. 1997, 46, 19–23. [Google Scholar] [CrossRef]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I. Role of c-myc in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Vadiveloo, P.; Keramidaris, E.; Morrison, W.; Stewart, A. Lipopolysaccharide-induced cell cycle arrest in macrophages occurs independently of nitric oxide synthase ii induction. BBA Rev. Can. 2001, 1539, 140–146. [Google Scholar] [CrossRef]
- Vadiveloo, P.K. Macrophages--proliferation, activation, and cell cycle proteins. J. Leukoc Biol 1999, 66, 579–582. [Google Scholar] [PubMed]
- Yamaguchi, H.; Maruyama, T.; Urade, Y.; Nagata, S. Immunosuppression via adenosine receptor activation by adenosine monophosphate released from apoptotic cells. Elife 2014, 3, e02172. [Google Scholar] [CrossRef] [PubMed]
- Pello, O.M.; Chèvre, R.; Laoui, D.; De Juan, A.; Lolo, F.; Andrés-Manzano, M.J.; Serrano, M.; Van Ginderachter, J.A.; Andrés, V. In vivo inhibition of c-myc in myeloid cells impairs tumor-associated macrophage maturation and pro-tumoral activities. PLoS One 2012, 7, e45399. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. C-myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell. Metab 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S.M. Glutamine metabolism in lymphocytes: Its biochemical, physiological and clinical importance. Q. J. Exp. Psychol. 1985, 70, 473–489. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell. 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Myc, metabolism, cell growth, and tumorigenesis. Csh. Perspect. Med. Med. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Droin, N.; Pinkoski, M. Activation-induced cell death in t cells. Immunol. Rev. 2003, 193, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Gerlach, C.; van Heijst, J.W. Mapping the life histories of t cells. Nat. Rev. Immunol. 2010, 10, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Green, D.R. The immune diet: Meeting the metabolic demands of lymphocyte activation. F1000 Biol Rep. 2012, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Green, D.R. Metabolic reprogramming and metabolic dependency in t cells. Immunol. Rev. 2012, 249, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Rathmell, J.C. Metabolic pathways in t cell fate and function. Trends Immunol. 2012, 33, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing cd8 t-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory cd4+ t cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in t cell activation and requires cd28-mediated akt-dependent and independent pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Farkash, E.A.; Gao, W.; Thompson, C.B. Il-7 enhances the survival and maintains the size of naive t cells. J. Immunol. 2001, 167, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The cd28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Verbist, K.C.; Wang, R.; Green, D.R. T cell metabolism and the immune response. Semin. Immunol. 2012, 24, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Verbist, K.; Wang, R.; Green, D.R. The relationship between metabolism and the autophagy machinery during the innate immune response. Cell. Metab. 2013, 17, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. Il-7 promotes glut1 trafficking and glucose uptake via stat5-mediated activation of akt to support t-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by erk/mapk during t lymphocyte activation. J. Immunol 2011, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. Hif1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of th17 and treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of t(h)17/t(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Tamas, P.; Hawley, S.A.; Clarke, R.G.; Mustard, K.J.; Green, K.; Hardie, D.G.; Cantrell, D.A. Regulation of the energy sensor amp-activated protein kinase by antigen receptor and ca2+ in t lymphocytes. J. Exp. Med. 2006, 203, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mtor kinase differentially regulates effector and regulatory t cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and function of mtor signalling in t cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Delgoffe, G.M. The mammalian target of rapamycin: Linking t cell differentiation, function, and metabolism. Immunity 2010, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Rathmell, J.C. The metabolic life and times of a t-cell. Immunol. Rev. 2010, 236, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl Acad Sci U S A 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (gls2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J. The transcription factor myc controls metabolic reprogramming upon t lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-myc suppression of mir-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Maciolek, J.A.; Pasternak, J.A.; Wilson, H.L. Metabolism of activated t lymphocytes. Curr. Opin. Immunol. 2014, 27, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int J. Biochem Cell. Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Moschou, P.N.; Roubelakis-Angelakis, K.A. Polyamines and programmed cell death. J. Exp. Bot. 2014, 65, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Bello-Fernandez, C.; Packham, G.; Cleveland, J.L. The ornithine decarboxylase gene is a transcriptional target of c-myc. Proc. Natl. Acad. Sci. USA 1993, 90, 7804–7808. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Paul, W.E. Peripheral cd4+ t-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010, 238, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of cd4+ t cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector th17 and regulatory t cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. Il-17 and th17 cells. Annu Rev. Immunol 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper cd4+ t cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P. The glucose transporter glut1 is selectively essential for cd4 t cell activation and effector function. Cell. Metabolism 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling th17 cell differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Mele, D.A.; Salmeron, A.; Ghosh, S.; Huang, H.-R.; Bryant, B.M.; Lora, J.M. Bet bromodomain inhibition suppresses th17-mediated pathology. J. Exp. Med. 2013, 210, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Bandukwala, H.S.; Gagnon, J.; Togher, S.; Greenbaum, J.A.; Lamperti, E.D.; Parr, N.J.; Molesworth, A.M.; Smithers, N.; Lee, K.; Witherington, J. Selective inhibition of cd4+ t-cell cytokine production and autoimmunity by bet protein and c-myc inhibitors. Proc. Natl. Acad. Sci. 2012, 109, 14532–14537. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shrestha, S.; Zeng, H.; Karmaus, P.W.; Neale, G.; Vogel, P.; Guertin, D.A.; Lamb, R.F.; Chi, H. T cell exit from quiescence and differentiation into th2 cells depend on raptor-mtorc1-mediated metabolic reprogramming. Immunity 2013, 39, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically baff-exposed b cells. J. Immunol 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Kobayashi, A.; Sakurai, D.; Kanno, Y.; Hase, H.; Takahashi, R.; Totsuka, Y.; Semenza, G.L.; Sitkovsky, M.V.; Kobata, T. Differentiation stage-specific requirement in hypoxia-inducible factor-1alpha-regulated glycolytic pathway during murine b cell development in bone marrow. J. Immunol 2010, 184, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Mambetsariev, N.; Lin, W.W.; Wallis, A.M.; Stunz, L.L.; Bishop, G.A. Traf3 deficiency promotes metabolic reprogramming in b cells. Sci. Rep. 2016, 6, 35349. [Google Scholar] [CrossRef] [PubMed]
- Doughty, C.A.; Bleiman, B.F.; Wagner, D.J.; Dufort, F.J.; Mataraza, J.M.; Roberts, M.F.; Chiles, T.C. Antigen receptor–mediated changes in glucose metabolism in b lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006, 107, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Jellusova, J.; Rickert, R.C. The pi3k pathway in b cell metabolism. Crit. Rev. Biochem. Mol. 2016, 51, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O'Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Galván-Peña, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front Immunol. 2014, 5, 420. [Google Scholar]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V. Hif-1α is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Tannahill, G.; Curtis, A.; Adamik, J.; Palsson-McDermott, E.; McGettrick, A.; Goel, G.; Frezza, C.; Bernard, N.; Kelly, B.; Foley, N. Succinate is an inflammatory signal that induces il-1 [bgr] through hif-1 [agr]. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. Hif transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Pantel, A.; Teixeira, A.; Haddad, E.; Wood, E.G.; Steinman, R.M.; Longhi, M.P. Direct type i ifn but not mda5/tlr3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly ic stimulation. PLoS Biol 2014, 12, e1001759. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages glucose transporter 1 (glut1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C. The sedoheptulose kinase carkl directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnanaprakasam, J.N.R.; Wang, R. MYC in Regulating Immunity: Metabolism and Beyond. Genes 2017, 8, 88. https://doi.org/10.3390/genes8030088
Gnanaprakasam JNR, Wang R. MYC in Regulating Immunity: Metabolism and Beyond. Genes. 2017; 8(3):88. https://doi.org/10.3390/genes8030088
Chicago/Turabian StyleGnanaprakasam, J.N. Rashida, and Ruoning Wang. 2017. "MYC in Regulating Immunity: Metabolism and Beyond" Genes 8, no. 3: 88. https://doi.org/10.3390/genes8030088
APA StyleGnanaprakasam, J. N. R., & Wang, R. (2017). MYC in Regulating Immunity: Metabolism and Beyond. Genes, 8(3), 88. https://doi.org/10.3390/genes8030088