
Possible Therapeutic Doses of Cannabinoid Type 1 Receptor Antagonist Reverses Key Alterations in Fragile X Syndrome Mouse Model





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drugs
2.3. Behavioral Tests
2.4. Electrophysiology
2.5. Statistical Analysis
3. Results
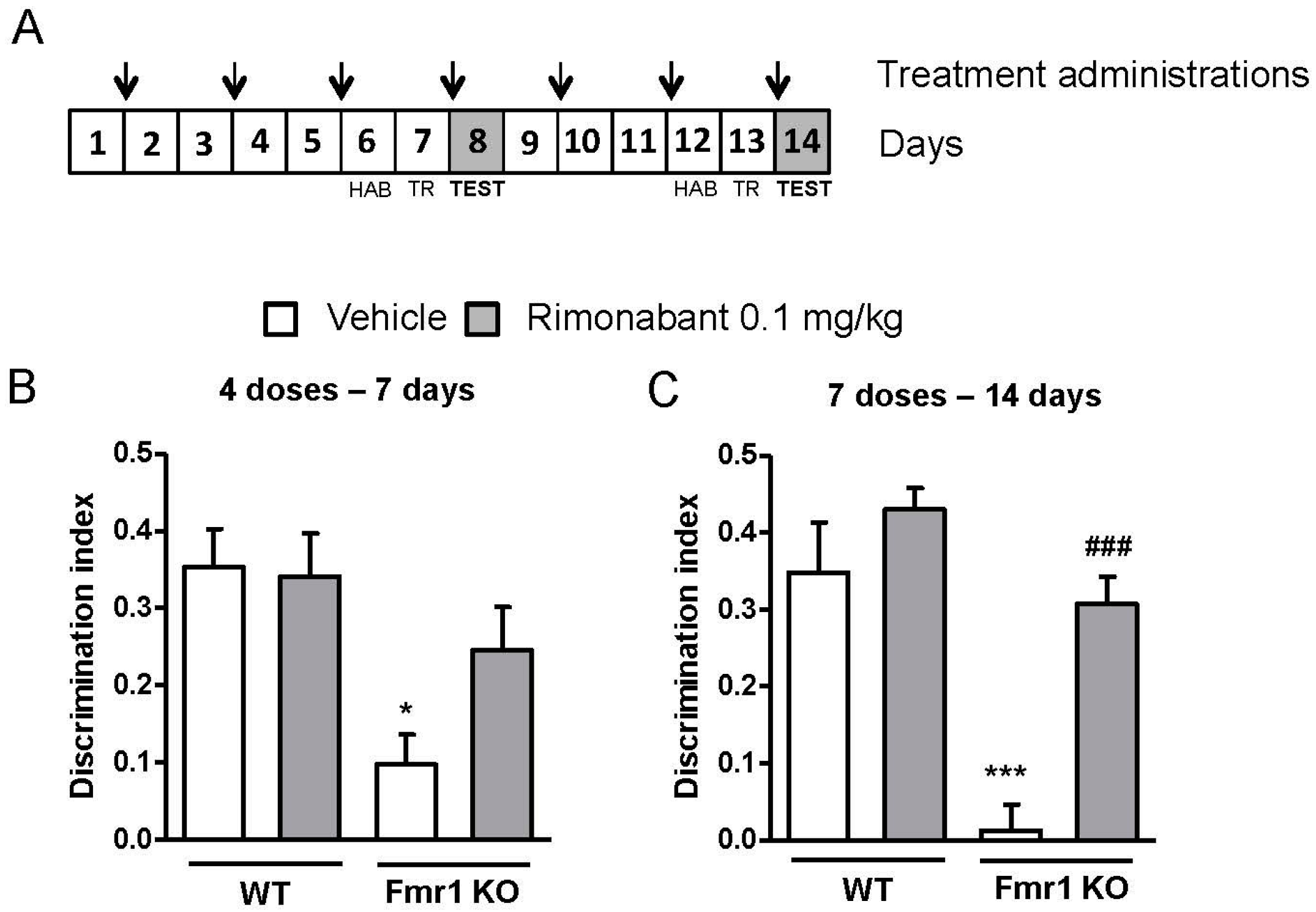
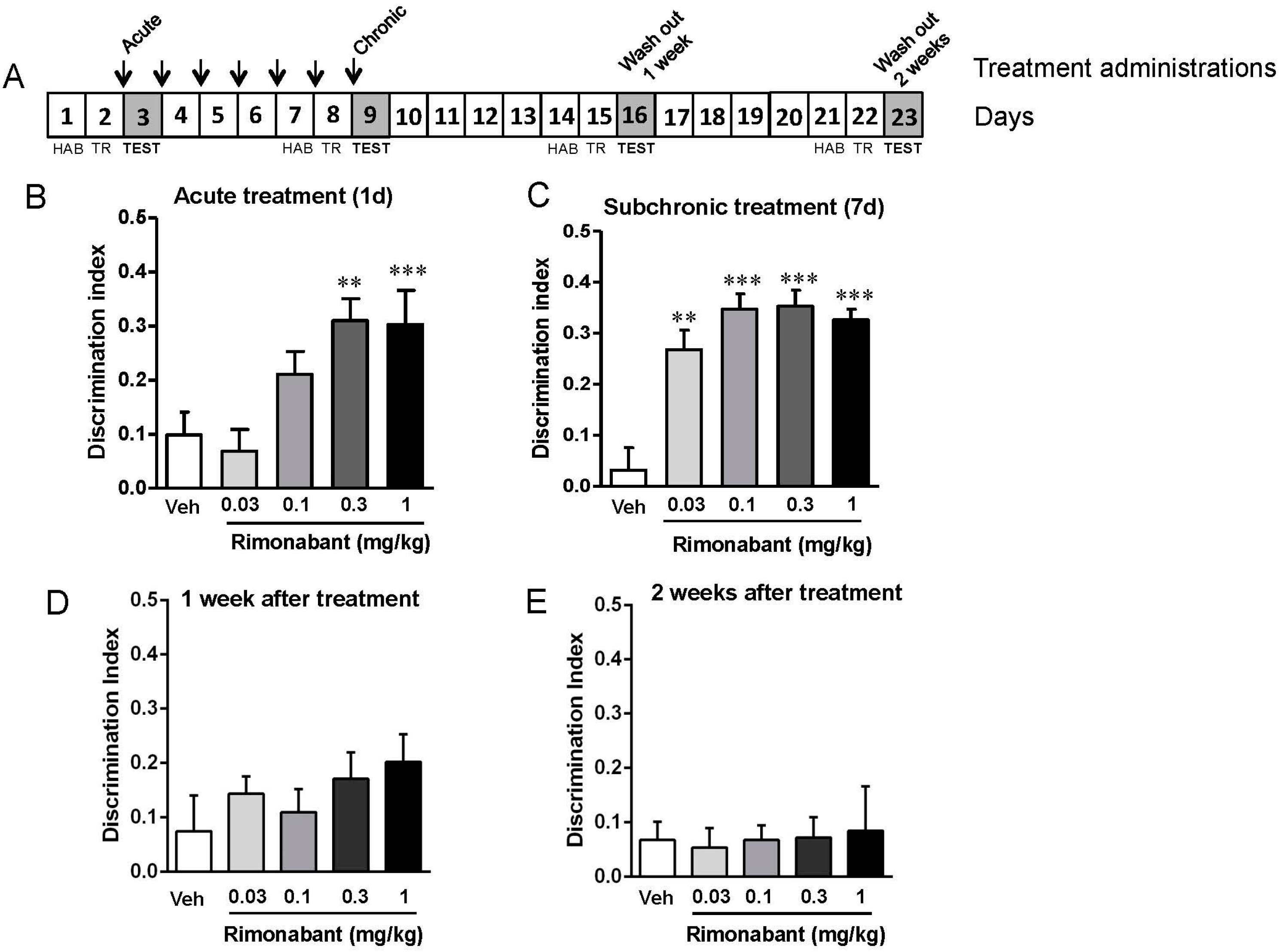
3.1. Low Doses of Rimonabant Ameliorate the Cognitive Impairment in Fmr1 KO Mice
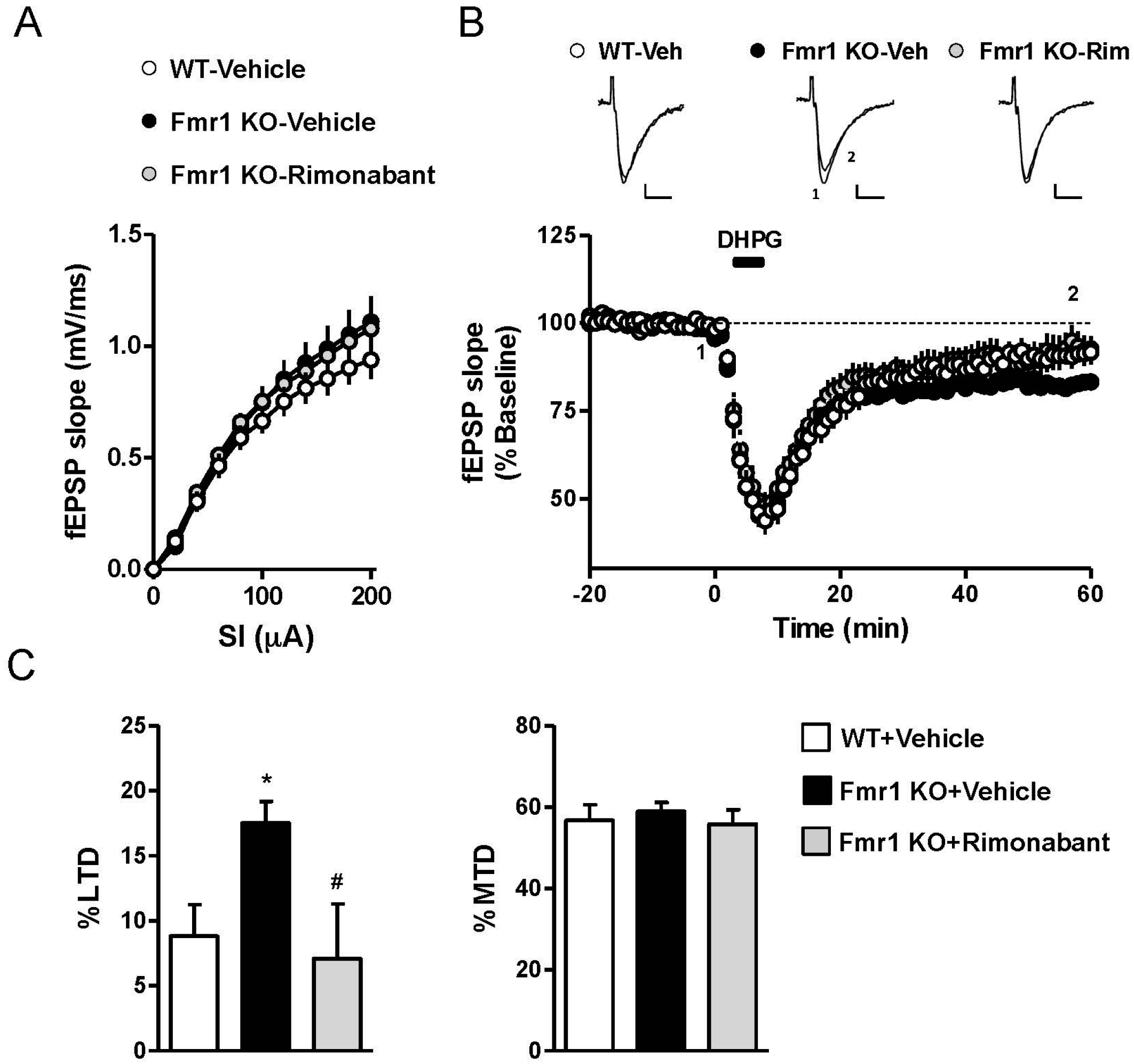
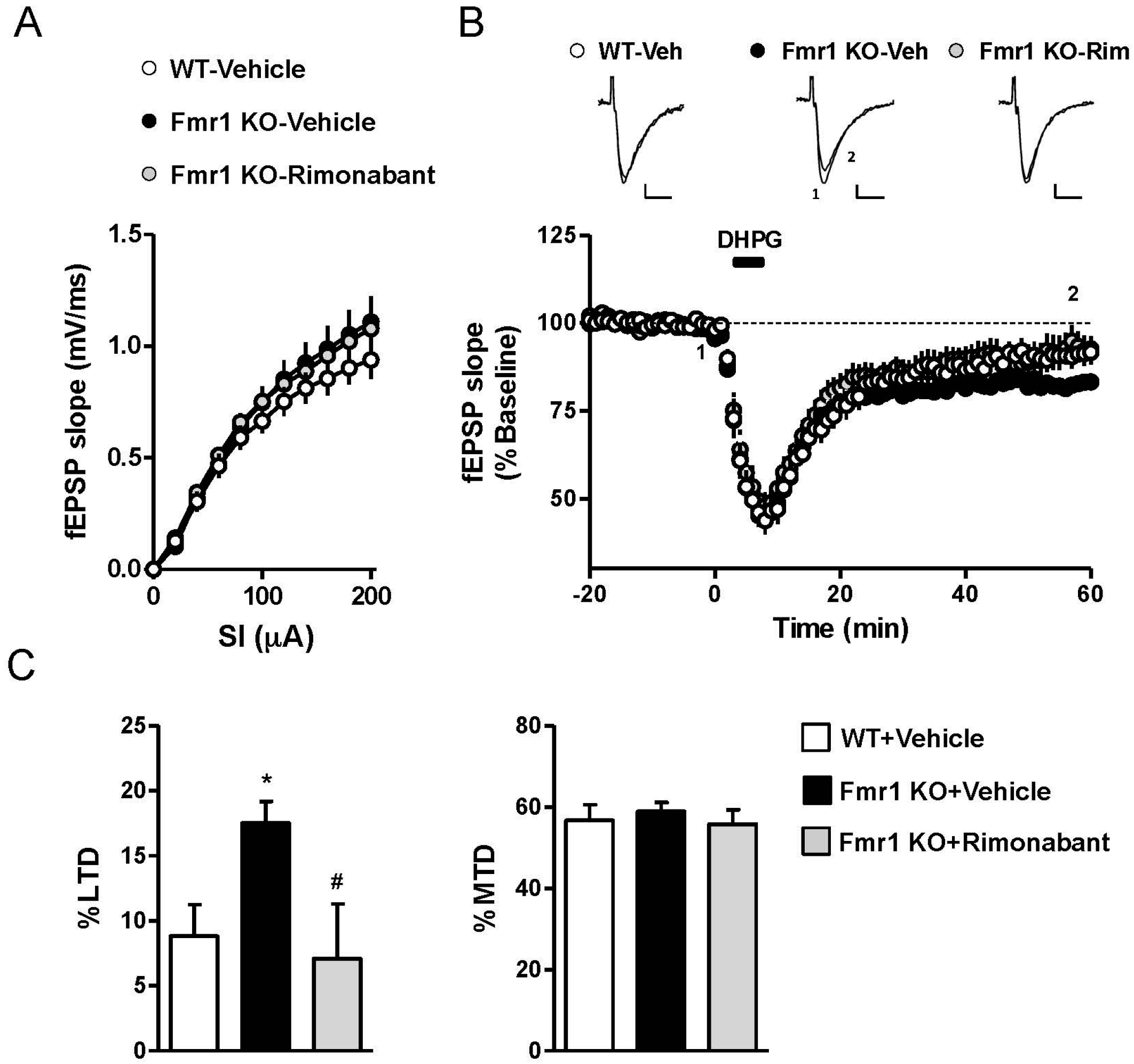
3.2. Rimonabant at a Low Dose Normalizes LTD in Fmr1 KO Mice
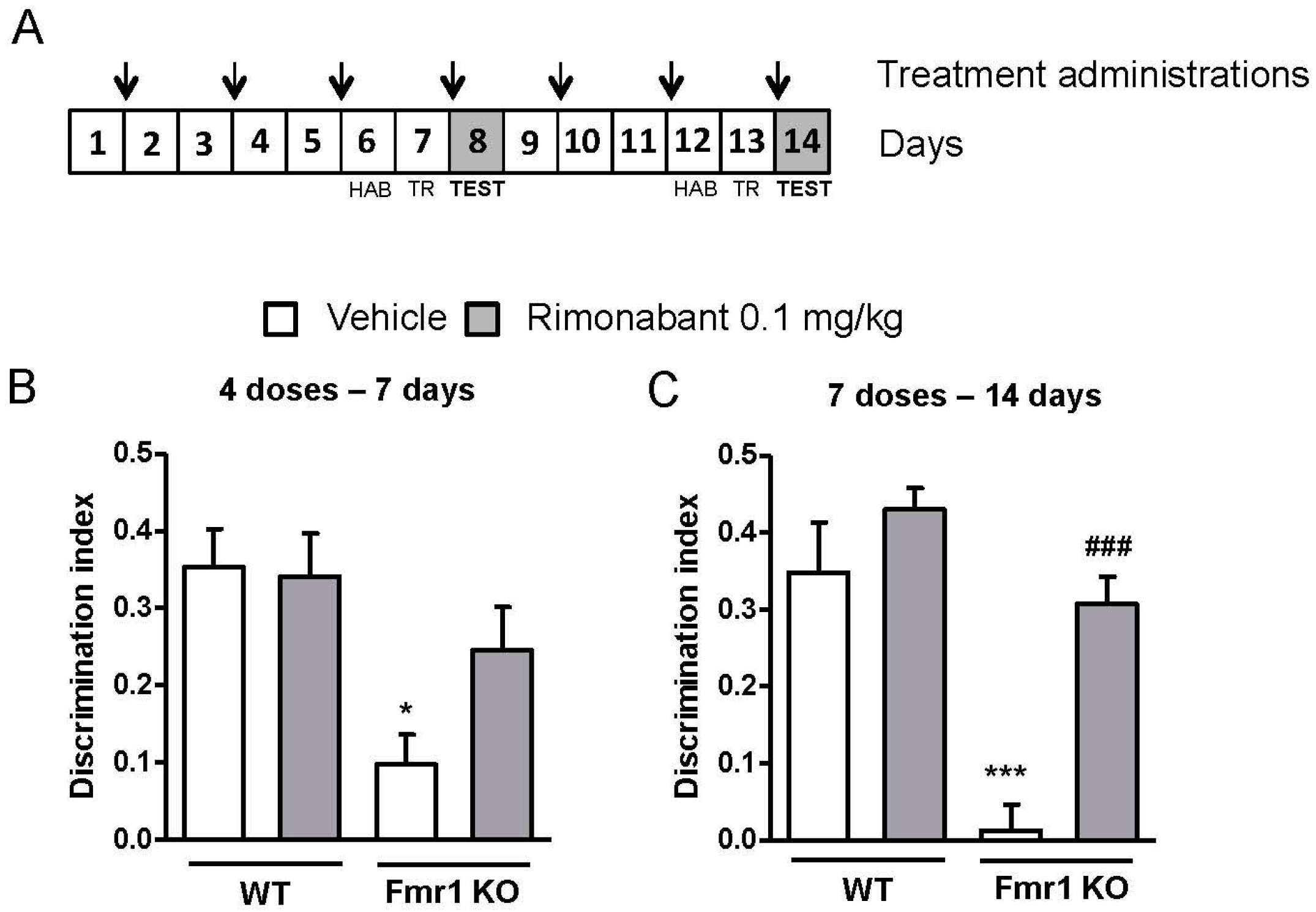
3.3. Discontinuous Treatment with a Low Dose of Rimonabant Reaches Normalization of the Cognitive Performance
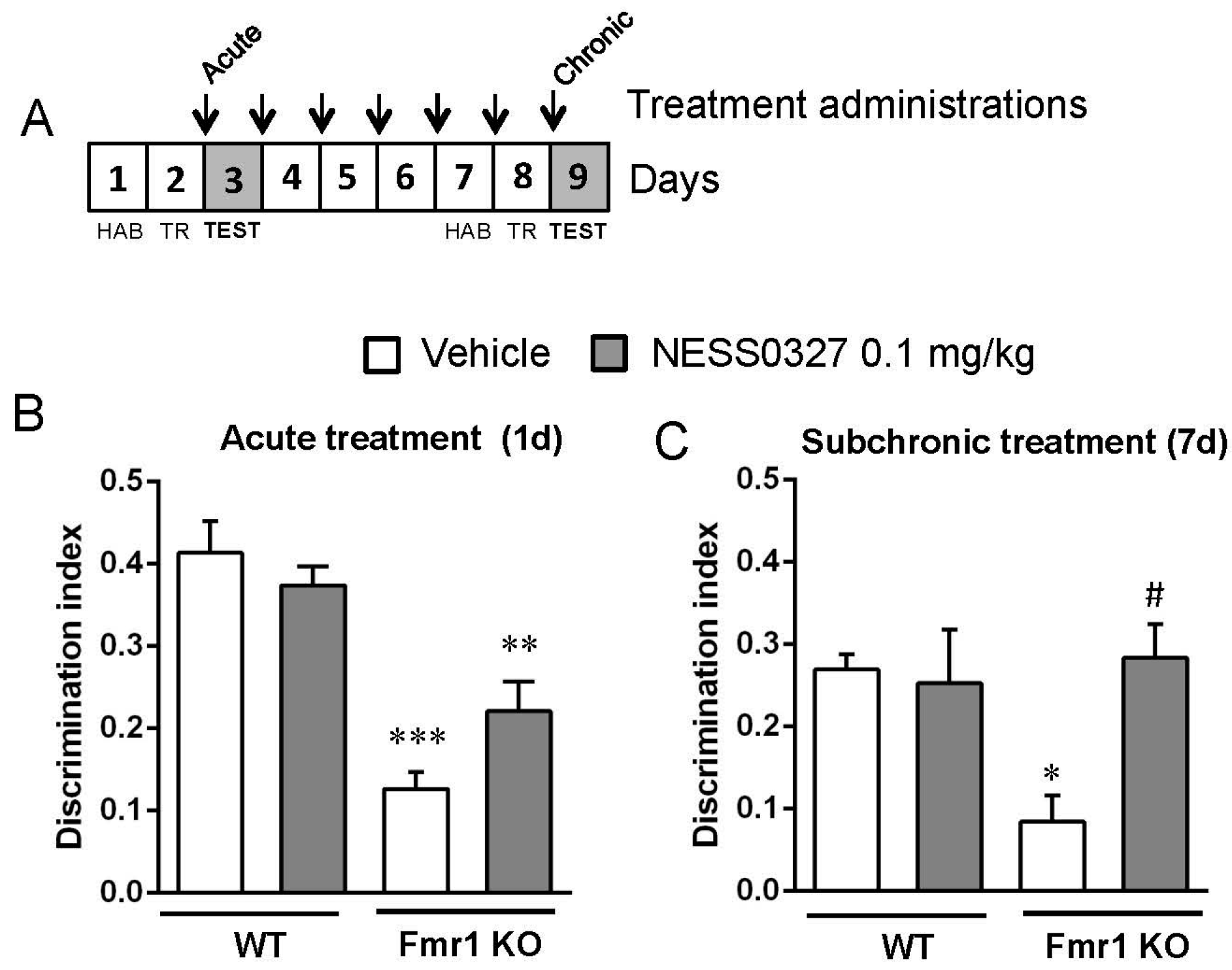
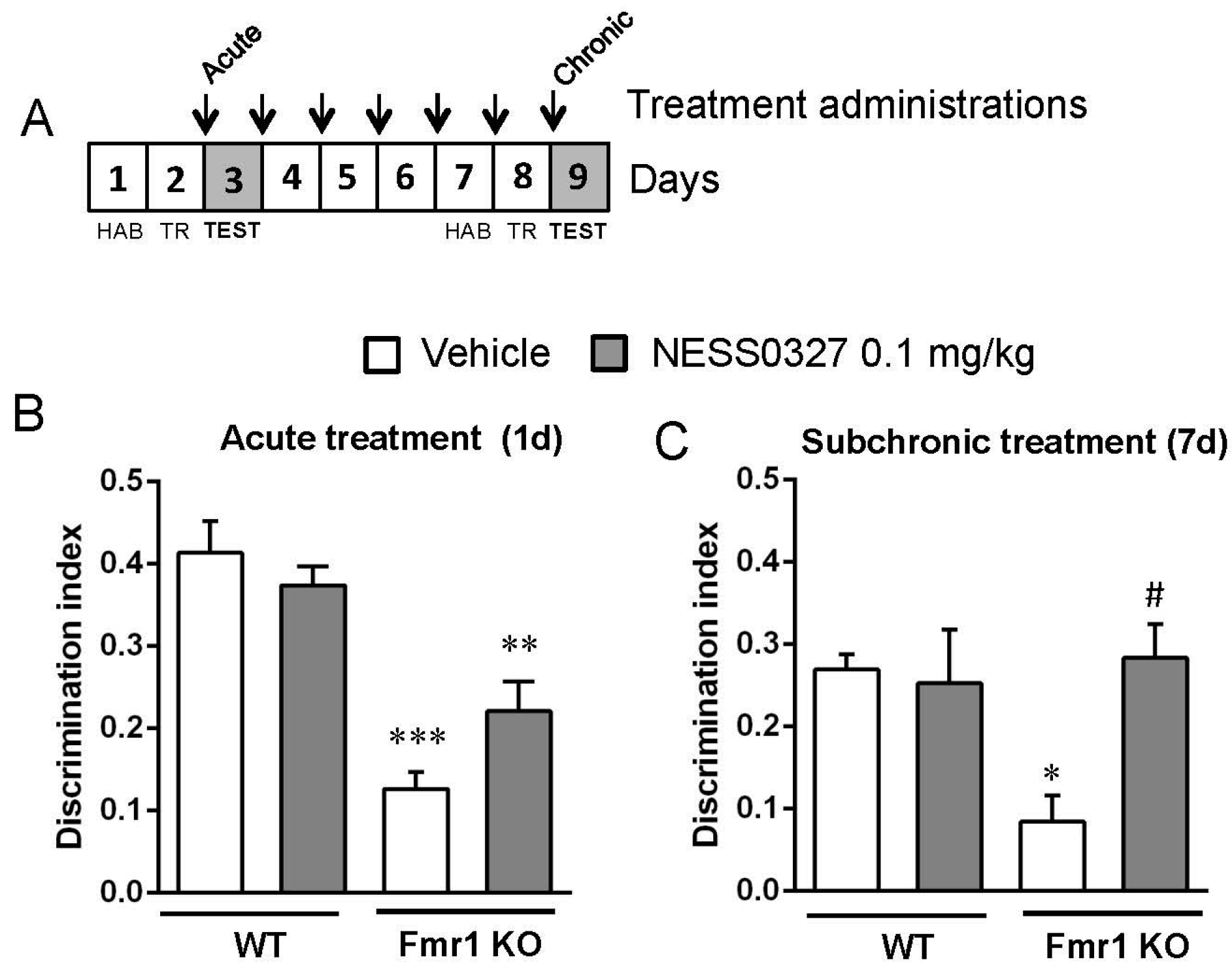
3.4. The CB1 Receptor Neutral Antagonist NESS0327 Also Has Beneficial Effects in Object-Recognition Memory
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Penagarikano, O.; Mulle, J.G.; Warren, S.T. The pathophysiology of fragile X syndrome. Annu. Rev. Genom. Hum. Genet. 2007, 8, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- O’Donnell, W.T.; Warren, S.T. A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 2002, 25, 315–338. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Warren, S.T. New insights into fragile X syndrome: From molecules to neurobehaviors. Trends Biochem. Sci. 2003, 28, 152–158. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef] [PubMed]
- D’Hulst, C.; Kooy, R.F. Fragile X syndrome: From molecular genetics to therapy. J. Med. Genet. 2009, 46, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.; Wijetunge, L.; Kinoshita, M.N.; Shumway, M.; Hammond, R.S.; Postma, F.R.; Brynczka, C.; Rush, R.; Thomas, A.; Paylor, R.; et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 2012, 4, 152ra128. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Lutz, B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 1999, 11, 4213–4225. [Google Scholar] [CrossRef] [PubMed]
- Varma, N.; Carlson, G.C.; Ledent, C.; Alger, B.E. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J. Neurosci. 2001, 21, RC188. [Google Scholar] [PubMed]
- Centonze, D.; Rossi, S.; Mercaldo, V.; Napoli, I.; Ciotti, M.T.; de Chiara, V.; Musella, A.; Prosperetti, C.; Calabresi, P.; Bernardi, G.; et al. Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol. Psychiatry 2008, 63, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [CrossRef] [PubMed]
- Suvrathan, A.; Hoeffer, C.A.; Wong, H.; Klann, E.; Chattarji, S. Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 11591–11596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Alger, B.E. Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome. J. Neurosci. 2010, 30, 5724–5729. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.M.; Sepers, M.; Henstridge, C.M.; Lassalle, O.; Neuhofer, D.; Martin, H.; Ginger, M.; Frick, A.; DiPatrizio, N.V.; Mackie, K.; et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 2012, 3, 1080. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Héaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Néliat, G.; Caput, D.; et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Rucker, D.; Padwal, R.; Li, S.K.; Curioni, C.; Lau, D.C. Long term pharmacotherapy for obesity and overweight: Updated meta-analysis. BMJ 2007, 335, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Landsman, R.S.; Burkey, T.H.; Consroe, P.; Roeske, W.R.; Yamamura, H.I. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur. J. Pharmacol. 1997, 334, R1–R2. [Google Scholar] [CrossRef]
- Silvestri, C.; di Marzo, V. Second generation CB1 receptor blockers and other inhibitors of peripheral endocannabinoid overactivity and the rationale of their use against metabolic disorders. Expert Opin. Investig. Drugs 2012, 21, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Ruiu, S.; Pinna, G.A.; Marchese, G.; Mussinu, J.M.; Saba, P.; Tambaro, S.; Casti, P.; Vargiu, R.; Pani, L. Synthesis and characterization of NESS 0327: A novel putative antagonist of the CB1 cannabinoid receptor. J. Pharmacol. Exp. Ther. 2003, 306, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Gomis-González, M.; Guegan, T.; Agustín-Pavón, C.; Pastor, A.; Mato, S.; Pérez-Samartín, A.; Matute, C.; de la Torre, R.; Dierssen, M.; et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat. Med. 2013, 19, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, H.; Dansie, L.E.; Hickmott, P.W.; Ethell, D.W.; Ethell, I.M. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J. Neurosci. 2014, 34, 9867–9879. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Burston, J.J.; Leggett, D.C.; Alekseeva, O.O.; Razdan, R.K.; Mahadevan, A.; Martin, B.R. CB1 cannabinoid receptor-mediated modulation of food intake in mice. Br. J. Pharmacol. 2005, 145, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Zádor, F.; Lénárt, N.; Csibrány, B.; Sántha, M.; Molnár, M.; Tuka, B.; Samavati, R.; Klivényi, P.; Vécsei, L.; Marton, A.; et al. Low dosage of rimonabant leads to anxiolytic-like behavior via inhibiting expression levels and G-protein activity of kappa opioid receptors in a cannabinoid receptor independent manner. Neuropharmacology 2015, 89, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005, 76, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Meye, F.J.; Trezza, V.; Vanderschuren, L.J.; Ramakers, G.M.; Adan, R.A. Neutral antagonism at the cannabinoid 1 receptor: A Safer treatment for obesity. Mol. Psychiatry 2013, 18, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Ledri, M.; Tóth, B.; Marchionni, I.; Henstridge, C.M.; Dudok, B.; Kenesei, K.; Barna, L.; Szabó, S.I.; Renkecz, T.; et al. Multiple forms of endocannabinoid and endovanilloid signaling regulate the tonic control of GABA release. J. Neurosci. 2015, 35, 10039–10057. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.J.; Irving, A.J.; Seabrook, G.R.; Jane, D.E.; Collingridge, G.L. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology 1997, 36, 1517–1532. [Google Scholar] [CrossRef]
- Sanderson, T.M.; Hogg, E.L.; Collingridge, G.L.; Corrêa, S.A. Hippocampal mGluR-LTD in health and disease: Focus on the p38 MAPK and ERK1/2 pathways. J. Neurochem. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Carpenter, R.L.; Ocain, T.D.; Bear, M.F. Mechanism-based approaches to treating fragile X. Pharmacol. Ther. 2010, 127, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Osterweil, E.K.; Chuang, S.C.; Chubykin, A.A.; Sidorov, M.; Bianchi, R.; Wong, R.K.; Bear, M.F. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 2013, 77, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Maldonado, R.; Ozaita, A. New insights into the molecular pathophysiology of fragile X syndrome and therapeutic perspectives from the animal model. Int. J. Biochem. Cell Biol. 2014, 53, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Martín-García, E.; Burokas, A.; Martín, M.; Berrendero, F.; Rubí, B.; Kiesselbach, C.; Heyne, A.; Gispert, J.D.; Millán, O.; Maldonado, R. Central and peripheral consequences of the chronic blockade of CB1 cannabinoid receptor with rimonabant or taranabant. J. Neurochem. 2010, 112, 1338–1351. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Sardone, L.M.; Lacivita, E.; Leopoldo, M.; Ciranna, L. Novel agonists for serotonin 5-HT7 receptors reverse metabotropic glutamate receptor-mediated long-term depression in the hippocampus of wild-type and Fmr1 KO mice, a model of Fragile X Syndrome. Front. Behav. Neurosci. 2015, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Picone, R.P.; Kendall, D.A. Minireview: From the bench, toward the clinic: Therapeutic opportunities for cannabinoid receptor modulation. Mol. Endocrinol. 2015, 29, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Till, S.M.; Asiminas, A.; Jackson, A.D.; Katsanevaki, D.; Barnes, S.A.; Osterweil, E.K.; Bear, M.F.; Chattarji, S.; Wood, E.R.; Wyllie, D.J.; et al. Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum. Mol. Genet. 2015, 24, 5977–5984. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomis-González, M.; Busquets-Garcia, A.; Matute, C.; Maldonado, R.; Mato, S.; Ozaita, A. Possible Therapeutic Doses of Cannabinoid Type 1 Receptor Antagonist Reverses Key Alterations in Fragile X Syndrome Mouse Model. Genes 2016, 7, 56. https://doi.org/10.3390/genes7090056
Gomis-González M, Busquets-Garcia A, Matute C, Maldonado R, Mato S, Ozaita A. Possible Therapeutic Doses of Cannabinoid Type 1 Receptor Antagonist Reverses Key Alterations in Fragile X Syndrome Mouse Model. Genes. 2016; 7(9):56. https://doi.org/10.3390/genes7090056
Chicago/Turabian StyleGomis-González, Maria, Arnau Busquets-Garcia, Carlos Matute, Rafael Maldonado, Susana Mato, and Andrés Ozaita. 2016. "Possible Therapeutic Doses of Cannabinoid Type 1 Receptor Antagonist Reverses Key Alterations in Fragile X Syndrome Mouse Model" Genes 7, no. 9: 56. https://doi.org/10.3390/genes7090056
APA StyleGomis-González, M., Busquets-Garcia, A., Matute, C., Maldonado, R., Mato, S., & Ozaita, A. (2016). Possible Therapeutic Doses of Cannabinoid Type 1 Receptor Antagonist Reverses Key Alterations in Fragile X Syndrome Mouse Model. Genes, 7(9), 56. https://doi.org/10.3390/genes7090056