
Towards a Better Molecular Diagnosis of FMR1-Related Disorders—A Multiyear Experience from a Reference Lab

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Molecular Diagnosis
2.2.1. Pre-Screening PCR
2.2.2. Follow-up Analysis for Samples with Uninformative Results
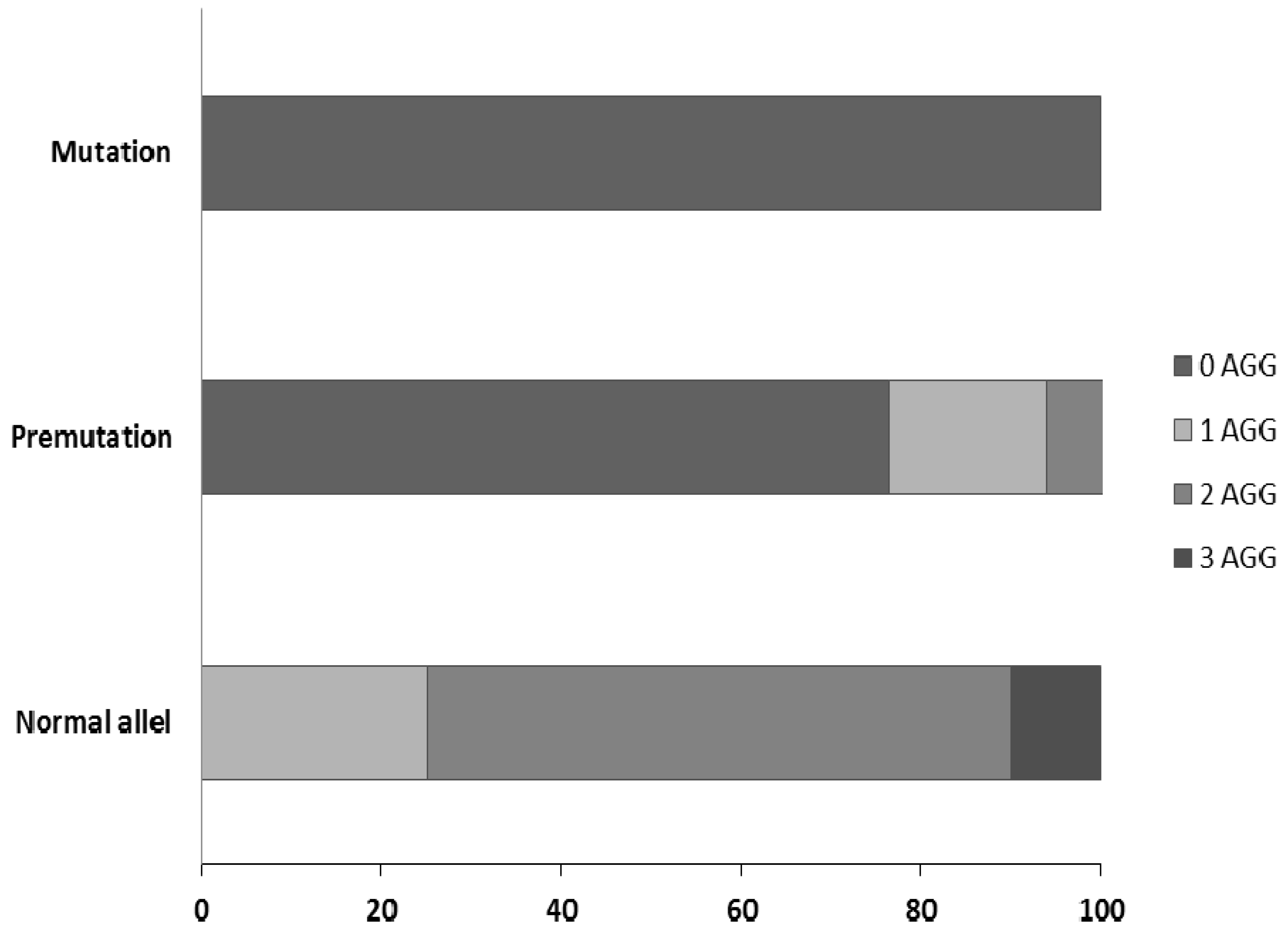
2.3. X Chromosome Inactivation Analysis
3. Results and Discussion
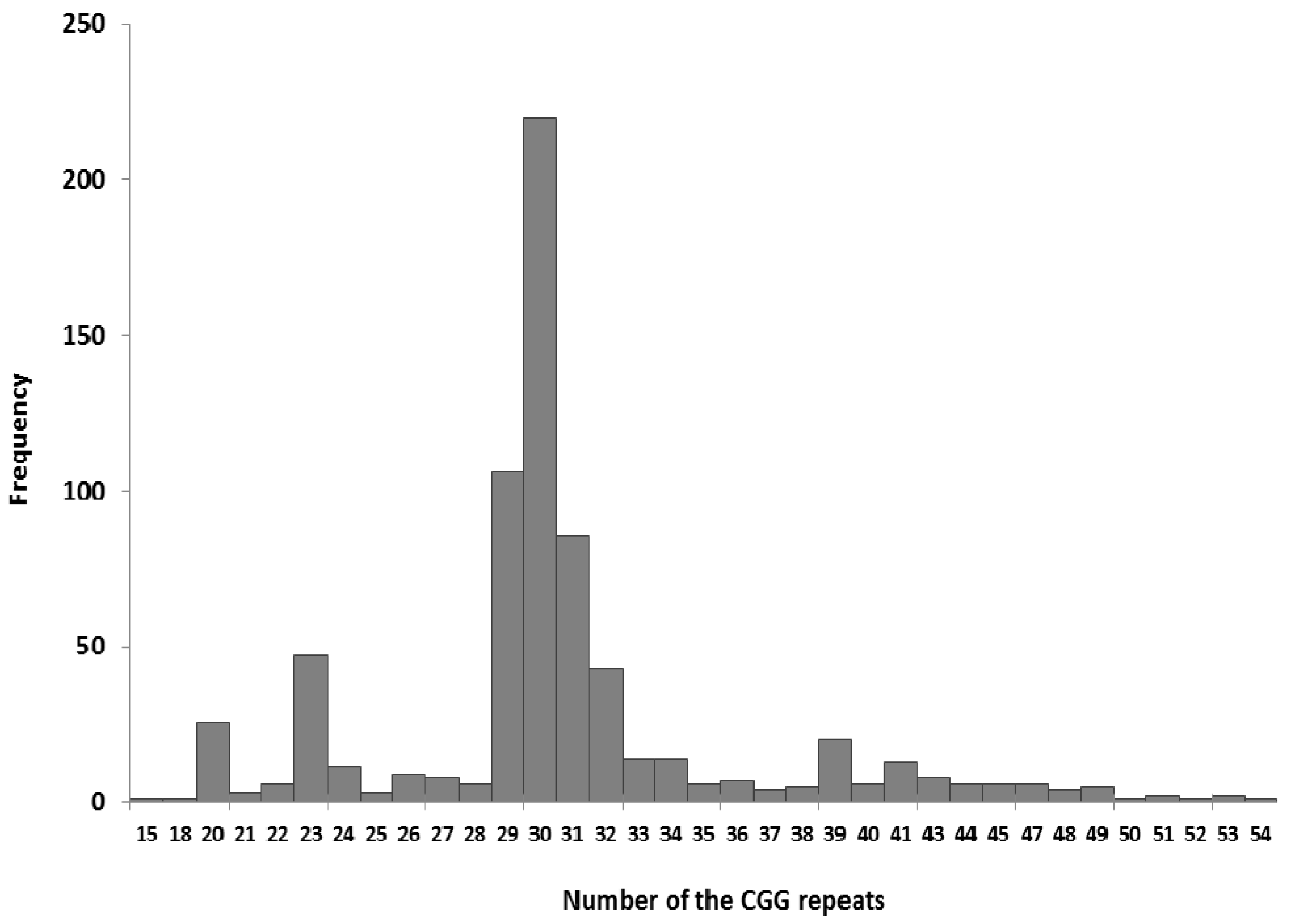
3.1. Testing the FMR1 Gene as a First-Line Test for Disturbances of Psychomotor Development
3.2. Testing for the FMR1 Gene in FXS Families
3.3. Prenatal Testing of the FMR1 Gene in FXS Families
3.4. Analysis of the FMR1 Gene in FXTAS and FXPOI Patients
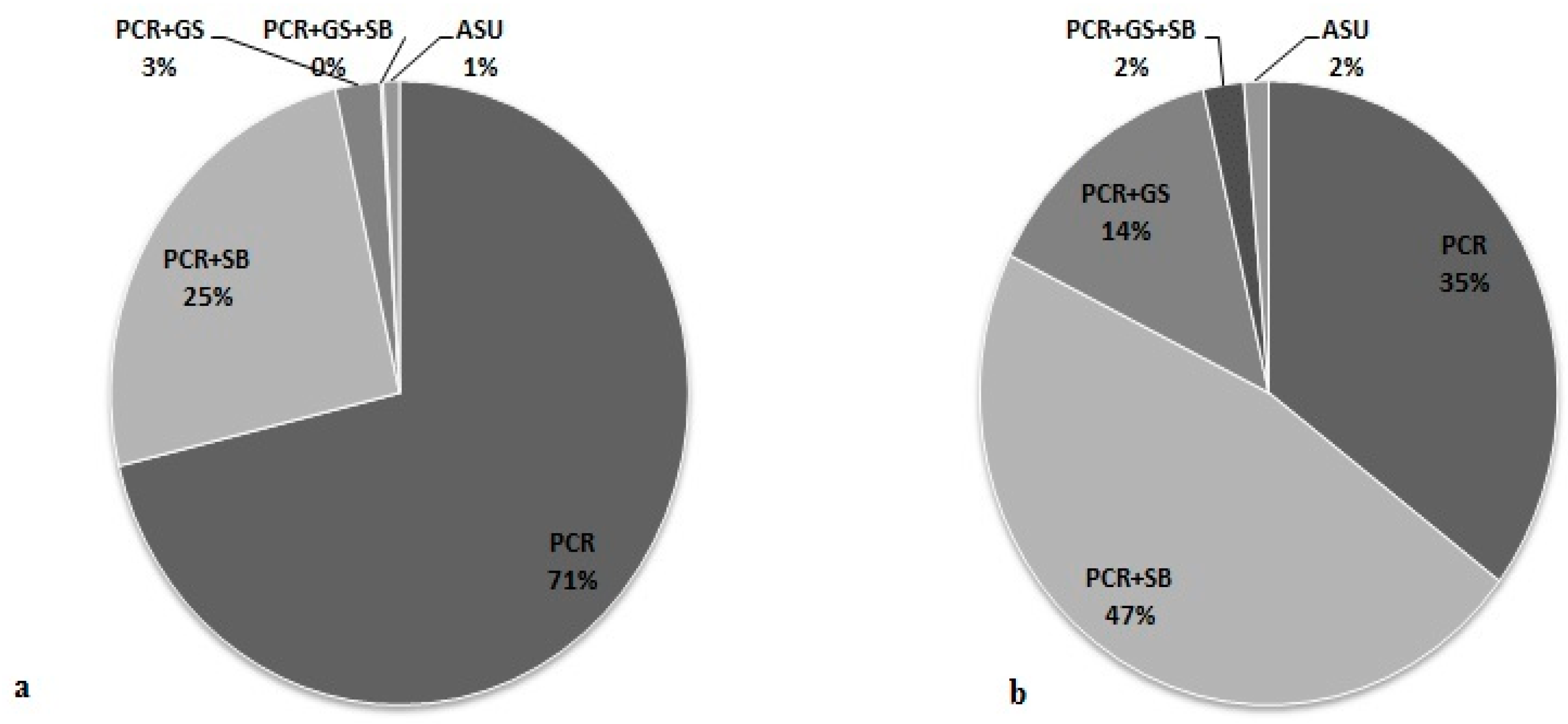
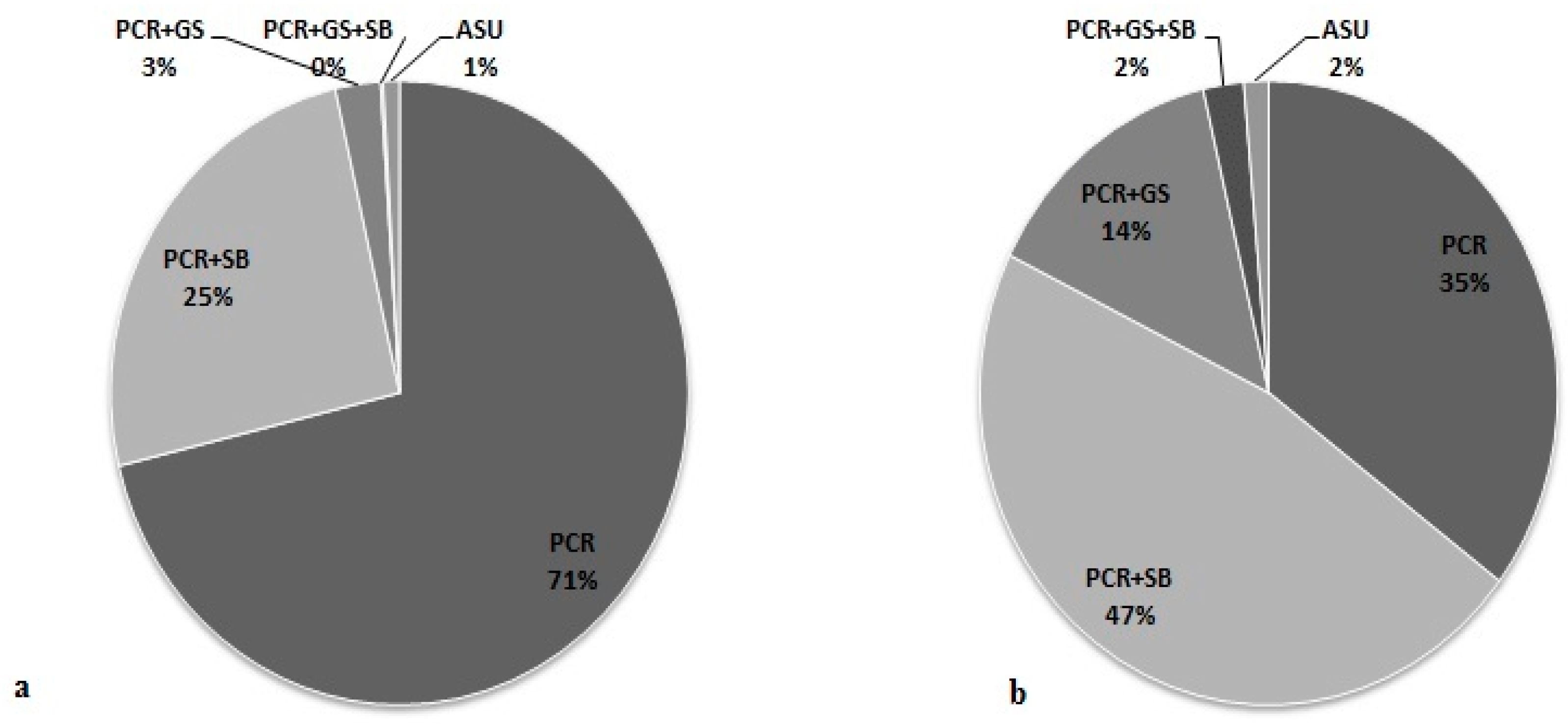
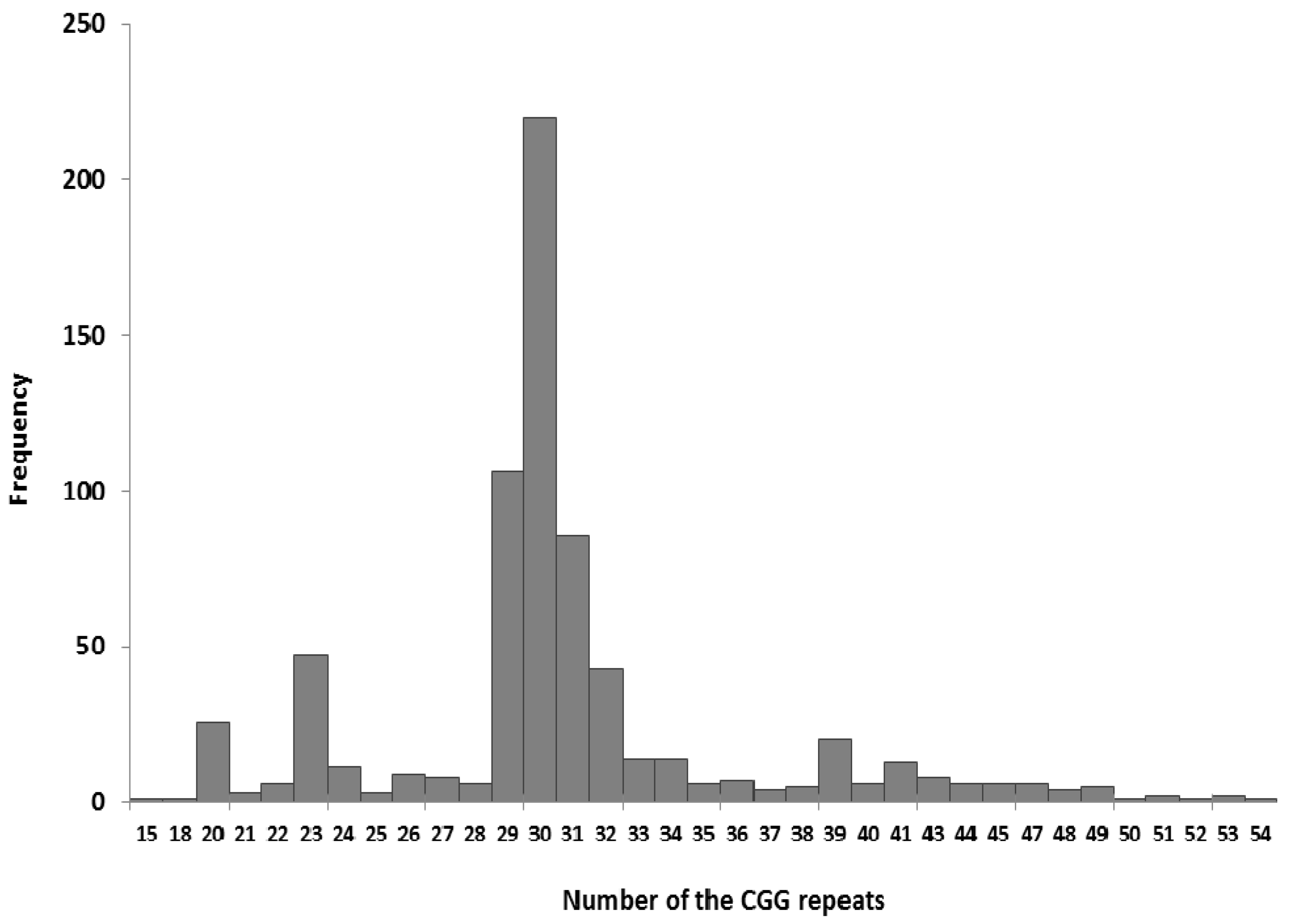
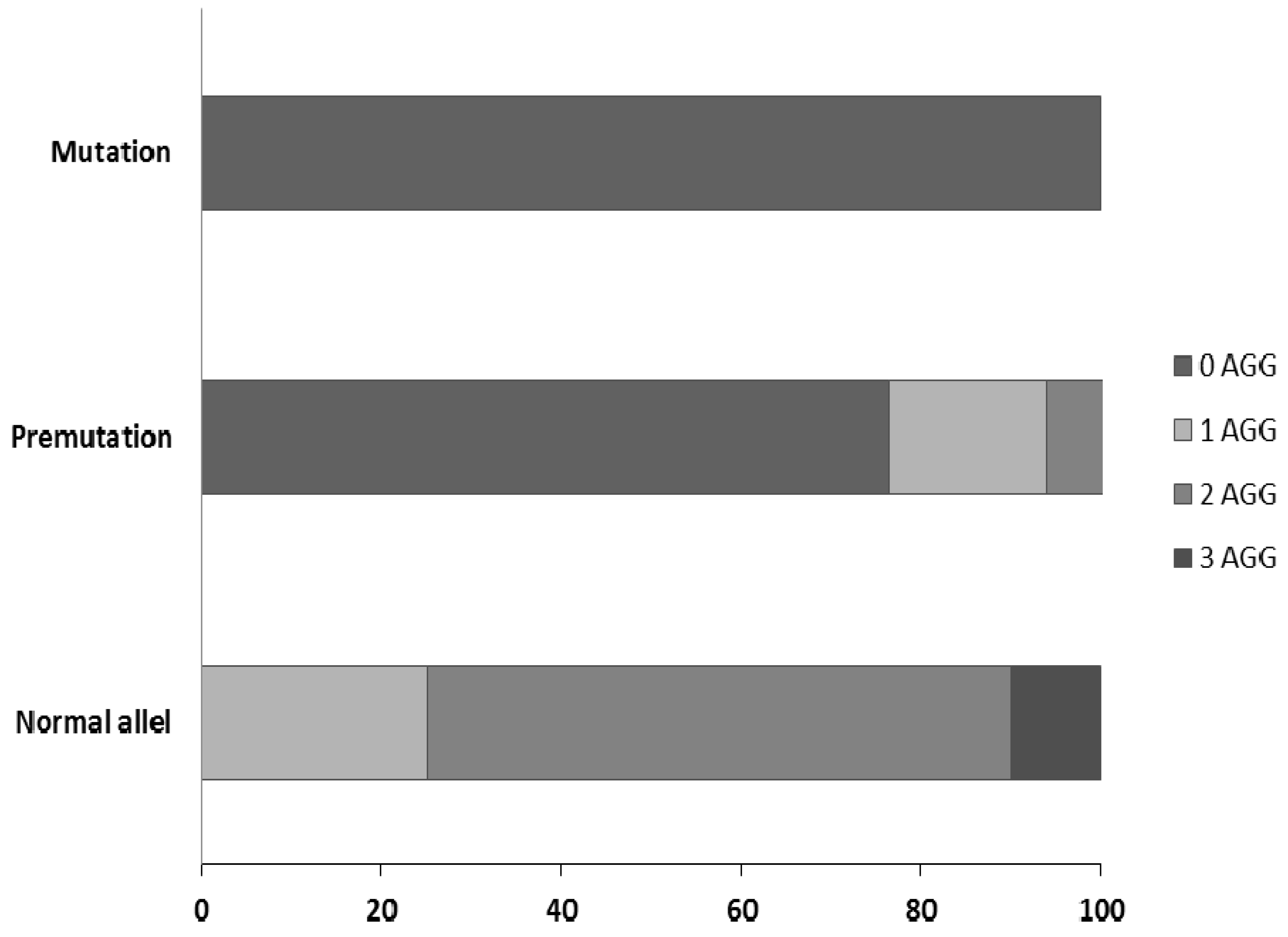
3.5. Effectiveness of Different Testing Protocols in FMR1 Gene Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Crawford, D.C.; Acuña, J.M.; Sherman, S.L. FMR1 and the fragile X syndrome: Human genome epidemiology review. Genet. Med. 2001, 3, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.C.; Knight, S.J.; Christodoulou, Z.; Grewal, P.K.; Fryns, J.P.; Davies, K.E. Origins of the fragile X syndrome mutation. J. Med. Genet. 1993, 30, 647–650. [Google Scholar] [CrossRef]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of Fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Bagni, C.; Klann, E. Molecular functions of the mammalian fragile X mental retardation protein: Insights into mental retardation and synaptic plasticity. In The Autisms; Powell, C.M., Monteggia, L.M., Eds.; Oxford University Press: Oxford, UK, 2013; pp. 126–139. [Google Scholar]
- Rzonca, S.O.; Gos, M.E. Rola białka FMRP w prawidłowym funkcjonowaniu organizmu oraz patogenezie zespołu łamliwego chromosomu X. Postęp. Biol. Komórki 2012, 39, 459–476. [Google Scholar]
- Biancalana, V.; Steinbach, P.; Glaeser, D. EMQN best Practice Guidelines for molecular analysis and reporting in Fragile X mental retardation syndrome (FXS), Fragile X-asociated tremor/ataxia syndrome (FXTAS), and Fragile X-associated primary ovarian insufficiency (FXPOI). Eur. Mol. Genet. Qual. Netw. 2013, 2, 1. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carvajal, I.; Posadas, B.L.; Pan, R.; Raske, C.; Hagerman, P.J.; Tassone, F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 2009, 11, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, G.E., Jr.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Bröndum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Bui, M.Q.; Hammersley, E.; Schneider, A.; Storey, E.; Stimpson, P.; Burgess, T.; Francis, D.; Slater, H.; Tassone, F.; et al. Psychological status in female carriers of premutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin. Genet. 2015, 87, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Iong, K.P.; Tong, T.H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012, 4, 100. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Rosero, C.A.; Hagerman, R.J. Fragile X spectrum disorders. Intractable Rare Dis. Res. 2014, 3, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Hagerman, P.J.; Leehey, M.A. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: Two faces of FMR1. Lancet 2007, 6, 45–55. [Google Scholar] [CrossRef]
- Murray, A.; Schoemaker, M.J.; Bennett, C.E.; Ennis, S.; Macpherson, J.N.; Jones, M.; Morris, D.H.; Orr, N.; Ashworth, A.; Jacobs, P.A.; et al. Population-based estimates of the prevalence of FMR1 expansion mutations in women with early menopause and primary ovarian insufficiency. Genet. Med. 2013, 16, 19–24. [Google Scholar] [CrossRef]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar] [PubMed]
- Lau, A.W.; Brown, C.J.; Penaherrera, M.; Langlois, S.; Kalousek, D.K.; Robinson, W.P. Skewed X-chromosome inactivation is common in fetuses or newborns associated with confined placental mosaicism. Am. J. Hum. Genet. 1997, 61, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Thurman, A.J.; McDuffie, A.; Hagerman, R.; Abbeduto, L. Psychiatric symptoms in boys with fragile X syndrome: A comparison with nonsyndromic autism spectrum disorder. Res. Dev. Disabil. 2014, 35, 1072–1086. [Google Scholar] [CrossRef]
- Tassone, F.; Choudhary, N.S.; Tassone, F.; Durbin-Johnson, B.; Hansen, R.; Hertz-Picciotto, I.; Pessah, I. Identification of expanded alleles of the FMR1 Gene in the Childhood Autism Risks from Genes and Environment (CHARGE) study. J. Autism Dev. Disord. 2013, 43, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Mazurczak, T.; Bocian, E.; Milewski, M.; Obersztyn, E.; Stanczak, H.; Bal, J.; Szamotulska, K.; Karwacki, M.W. Frequency of Fra X syndrome among institutionalized mentally retarded males in Poland. Am. J. Med. Genet. 1996, 64, 184–186. [Google Scholar] [CrossRef]
- Esposito, G.; Ruggiero, R.; Savarese, G.; Savarese, M.; Tremolaterra, M.R.; Salvatore, F.; Carsana, A. A 15-year case-mix experience for fragile X syndrome molecular diagnosis and comparison between conventional and alternative techniques leading to a novel diagnostic procedure. Clin. Chim. Acta 2013, 417, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Huggins, R.M.; Bui, Q.M.; Taylor, A.K.; Hagerman, R.J. Relationship of deficits of FMR1 gene specific protein with physical phenotype of fragile X males and females in pedigrees: A new perspective. Am. J. Med. Genet. A 2003, 118, 127–134. [Google Scholar] [CrossRef]
- Han, X.D.; Powell, B.R.; Phalin, J.L.; Chehab, F.F. Mosaicism for a full mutation, premutation, and deletion of the CGG repeats results in 22% FMRP and elevated FMR1 mRNA levels in a high-functioning fragile X male. Am. J. Med. Genet. 2006, 140, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- The Human Gene Mutataion Database. Available online: http://www.hgmd.org/ (accessed on 14 August 2014).
- Pretto, D.I.; Mendoza-Morales, G.; Lo, J.; Cao, R.; Hadd, A.; Latham, G.J.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 2014, 51, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Heine-Suñer, D.; Torres-Juan, L.; Morla, M.; Busquets, X.; Barcelo, F.; Pico, G.; Bonilla, L.; Govea, N.; Bernues, M.; Rosell, J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: A female member with complete inactivation of the functional X chromosome. Am. J. Med. Genet. A 2003, 122, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Inaba, Y.; Shi, E.Z.; Skinner, C.; Bui, Q.M.; Francis, D.; Amor, D.J.; Hopper, J.L.; Loesch, D.Z.; Hagerman, R.J.; et al. Relationships between age and epi-genotype of the FMR1 exon 1/intron 1 boundary are consistent with non-random X-chromosome inactivation in FM individuals, with the selection for the unmethylated state being most significant between birth and puberty. Hum. Mol. Genet. 2013, 22, 1516–1524. [Google Scholar] [CrossRef]
- Ferreira, S.I.; Pires, L.M.; Ferrro, J.; Sa, J.; Serra, A.; Careira, M. Mosaicism for FMR1 gene full mutation and intermediate allele in a female foetus: A postzygotic retraction event. Gene 2013, 527, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Bontekoe, C.J.; Severijnen, L.A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Wittenberger, M.D.; Hagerman, R.J.; Sherman, S.L.; McConkie-Rosell, A.; Welt, C.K.; Rebar, R.W.; Corrigan, E.C.; Simpson, J.L.; Nelson, L.M. The FMR1 premutation and reproduction. Fertil. Steril. 2007, 87, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X Premutation Tremor/Ataxia Syndrome: Molecular, Clinical, and Neuroimaging Correlates. Am. J. Hum. Genet. 2003, 72, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.; Pletcher, B.A.; Driscoll, D.A. Fragile X syndrome: Diagnostic and carrier testing. Genet. Med. 2005, 7, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Rajkiewicz, M.; Szlendak-Sauer, K.; Sulek, A.; Gawlik-Zawislak, S.; Krysa, W.; Radowicki, S.; Zaremba, J. A molecular and cytogenetic investigation of FMR1 gene premutations in Polish patients with primary ovarian insufficiency. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 155, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Pastore, L.M.; Johnson, J. The FMR1 gene, infertility, and reproductive decision-making: A review. Front. Genet. 2014, 5, 195. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. J. Am. Med. Assoc. 2004, 291, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Leedey, M.A.; Hagerman, R.J.; Beckett, L.A.; Hagerman, P.J. Size bias of fragile X premutation alleles in late-onset movement disorders. J. Med. Genet. 2006, 43, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Apartis, E.; Blancher, A.; Meissner, W.G.; Guyant-Maréchal, L.; Maltête, D.; de Broucker, T.; Legrand, A.P.; Bouzenada, H.; Thanh, H.T.; Sallansonnet-Froment, M.; et al. FXTAS: New insights and the need for revised diagnostic criteria. Neurology 2012, 79, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.A.; Steenblock, K.J.; Thibodeau, S.N.; Lindor, N.M. Premutations in the FMR1 gene are uncommon in men undergoing genetic testing for spinocerebellar ataxia. J. Neurogenet. 2008, 22, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Rajkiewicz, M.; Sułek-Piatkowska, A.; Krysa, W.; Zdzienicka, E.; Szirkowiec, W.; Zaremba, J. Screening for premutation in the FMR1 gene in male patients suspected of spinocerebellar ataxia. Neurol. Neurochir. Pol. 2008, 42, 497–504. [Google Scholar] [PubMed]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J.; et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hagerman, P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013, 12, 786–798. [Google Scholar] [CrossRef]
- Latham, G.J.; Coppinger, J.; Hadd, A.G.; Nolin, S.L. The role of AGG interruptions in fragile X repeat expansions: A twenty-year perspective. Front. Genet. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Full Mutation | Premutation | Normal | Total | |
|---|---|---|---|---|
| N = 636 | N = 389 | N = 8160 | N = 9185 | |
| FXS/ID probands | 406 | 30 | 6969 | 7405 |
| Males | 385 | 18 | 5680 | 6083 |
| Females | 21 | 12 | 1289 | 1322 |
| Symptomatic relatives testing | 119 | 19 | 491 | 629 |
| Males | 102 | 1 | 330 | 337 |
| Females | 17 | 8 | 161 | 192 |
| Brothers | 73 | 3 | 201 | 70 |
| Sisters | 9 | 0 | 57 | 68 |
| Mothers | 1 | 4 | 44 | 65 |
| Other relatives | 36 | 3 | 189 | 334 |
| Asymptomatic carrier testing | 104 | 333 | 638 | 1075 |
| Males | 0 | 27 | 226 | 384 |
| Females | 104 | 306 | 412 | 840 |
| Mothers | 38 | 229 | 215 | 449 |
| Sisters | 47 | 13 | 97 | 141 |
| Brothers | 0 | 3 | 89 | 92 |
| Fathers | 0 | 2 | 17 | 19 |
| Grandfathers | 0 | 14 | 7 | 22 |
| Aunts | 5 | 40 | 28 | 73 |
| Other relatives | 14 | 33 | 185 | 242 |
| Prenatal diagnosis | 7 | 0 | 15 | 22 * |
| 2—somatic mosaicism | ||||
| Male | 3 | 0 | 10 | 13 |
| Female | 4 | 0 | 5 | 9 |
| FXTAS | 0 | 3 | 8 | 11 |
| FXPOI | 0 | 4 | 39 | 43 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzońca, S.O.; Gos, M.; Szopa, D.; Sielska-Rotblum, D.; Landowska, A.; Szpecht-Potocka, A.; Milewski, M.; Czekajska, J.; Abramowicz, A.; Obersztyn, E.; et al. Towards a Better Molecular Diagnosis of FMR1-Related Disorders—A Multiyear Experience from a Reference Lab. Genes 2016, 7, 59. https://doi.org/10.3390/genes7090059
Rzońca SO, Gos M, Szopa D, Sielska-Rotblum D, Landowska A, Szpecht-Potocka A, Milewski M, Czekajska J, Abramowicz A, Obersztyn E, et al. Towards a Better Molecular Diagnosis of FMR1-Related Disorders—A Multiyear Experience from a Reference Lab. Genes. 2016; 7(9):59. https://doi.org/10.3390/genes7090059
Chicago/Turabian StyleRzońca, Sylwia Olimpia, Monika Gos, Daniel Szopa, Danuta Sielska-Rotblum, Aleksandra Landowska, Agnieszka Szpecht-Potocka, Michał Milewski, Jolanta Czekajska, Anna Abramowicz, Ewa Obersztyn, and et al. 2016. "Towards a Better Molecular Diagnosis of FMR1-Related Disorders—A Multiyear Experience from a Reference Lab" Genes 7, no. 9: 59. https://doi.org/10.3390/genes7090059
APA StyleRzońca, S. O., Gos, M., Szopa, D., Sielska-Rotblum, D., Landowska, A., Szpecht-Potocka, A., Milewski, M., Czekajska, J., Abramowicz, A., Obersztyn, E., Maciejko, D., Mazurczak, T., & Bal, J. (2016). Towards a Better Molecular Diagnosis of FMR1-Related Disorders—A Multiyear Experience from a Reference Lab. Genes, 7(9), 59. https://doi.org/10.3390/genes7090059