
DNA Polymerases λ and β: The Double-Edged Swords of DNA Repair


Abstract
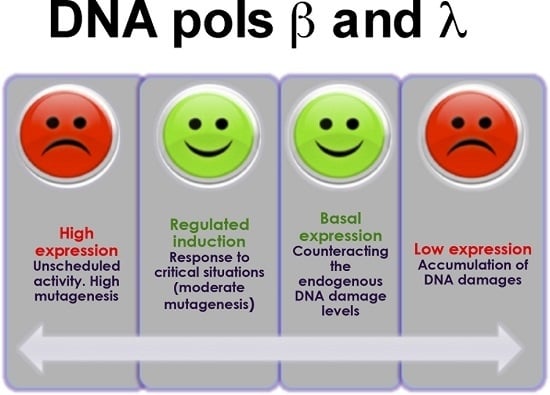
:
1. Introduction
2. Pol β and λ in the Base Excision Repair Pathway
3. Pol β and λ in the Translesion Synthesis Pathway
3.1. Bypass of the 7,8-Dihydro-8-Oxoguanine (8-oxo-G) Lesion
3.2. Bypass of Abasic Sites and the 2-Deoxyribonolactone Lesion
3.3. Pol δ-Interacting Protein 2 as an Auxiliary Factor for Pol λ during TLS
4. Pol β and λ and the Incorporation of Ribonucleotides in the Genome
5. Pol β and λ in Specialized Forms of DNA Double Stand Break Repair
6. Pol β and λ Roles in Genetic Instability
6.1. Pol λ
6.2. Pol β
7. Pols β and λ as Targets for Anticancer Chemotherapy
7.1. Pol β Natural Inhibitors
7.2. Pol β Synthetic Small Molecule Inhibitors
7.3. Pol λ Natural Inhibitors
7.4. Pol λ Synthetic Small Molecule Inhibitors
7.5. Dual Pols λ and β Natural Inhibitors
8. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Khoronenkova, S.V.; Dianov, G.L. The emerging role of mule and ARF in the regulation of base excision repair. FEBS Lett. 2011, 585, 2831–2835. [Google Scholar] [PubMed]
- Parsons, J.L.; Dianova, I.I.; Khoronenkova, S.V.; Edelmann, M.J.; Kessler, B.M.; Dianov, G.L. Usp47 is a deubiquitylating enzyme that regulates base excision repair by controlling steady-state levels of DNA polymerase β. Mol. Cell 2011, 41, 609–615. [Google Scholar] [PubMed]
- Frouin, I.; Toueille, M.; Ferrari, E.; Shevelev, I.; Hubscher, U. Phosphorylation of human DNA polymerase λ by the cyclin-dependent kinase cdk2/cyclin a complex is modulated by its association with proliferating cell nuclear antigen. Nucl. Acids Res. 2005, 33, 5354–5361. [Google Scholar] [PubMed]
- Markkanen, E.; Hübscher, U.; van Loon, B. Regulation of oxidative DNA damage repair: The adenine: 8-oxo-guanine problem. Cell Cycle 2012, 11, 1070–1075. [Google Scholar] [PubMed]
- Zucca, E.; Bertoletti, F.; Wimmer, U.; Ferrari, E.; Mazzini, G.; Khoronenkova, S.; Grosse, N.; van Loon, B.; Dianov, G.; Hubscher, U.; et al. Silencing of human DNA polymerase λ causes replication stress and is synthetically lethal with an impaired S phase checkpoint. Nucl. Acids Res. 2013, 41, 229–241. [Google Scholar] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [PubMed]
- Moon, A.F.; Garcia-Diaz, M.; Batra, V.K.; Beard, W.A.; Bebenek, K.; Kunkel, T.A.; Wilson, S.H.; Pedersen, L.C. The X family portrait: Structural insights into biological functions of X family polymerases. DNA Repair 2007, 6, 1709–1725. [Google Scholar] [PubMed]
- Bebenek, K.; Pedersen, L.C.; Kunkel, T.A. Structure-function studies of DNA polymerase λ. Biochemistry 2014, 53, 2781–2792. [Google Scholar] [PubMed]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucl. Acids Res. 2013, 41, 3483–3490. [Google Scholar] [PubMed]
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [PubMed]
- Beard, W.A.; Wilson, S.H. Structure and mechanism of DNA polymerase β. Chem. Rev. 2006, 106, 361–382. [Google Scholar] [PubMed]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244. [Google Scholar] [PubMed]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.-K.; van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial roles for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [PubMed]
- Maga, G.; Villani, G.; Crespan, E.; Wimmer, U.; Ferrari, E.; Bertocci, B.; Hubscher, U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature 2007, 447, 606–608. [Google Scholar] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [PubMed]
- Hübscher, U.; Maga, G. DNA replication and repair bypass machines. Curr. Opin. Chem. Biol. 2011, 15, 627–635. [Google Scholar] [PubMed]
- Amoroso, A.; Crespan, E.; Wimmer, U.; Hubscher, U.; Maga, G. DNA polymerases and oxidative damage: Friends or foes? Curr. Mol. Pharmacol. 2008, 1, 162–170. [Google Scholar] [PubMed]
- Maga, G.; Crespan, E.; Wimmer, U.; van Loon, B.; Amoroso, A.; Mondello, C.; Belgiovine, C.; Ferrari, E.; Locatelli, G.; Villani, G.; et al. Replication protein a and proliferating cell nuclear antigen coordinate DNA polymerase selection in 8-oxo-guanine repair. Proc. Natl. Acad. Sci. USA 2008, 105, 20689–20694. [Google Scholar] [PubMed]
- Malyarchuk, S.; Castore, R.; Harrison, L. DNA repair of clustered lesions in mammalian cells: Involvement of non-homologous end-joining. Nucl. Acids Res. 2008, 36, 4872–4882. [Google Scholar] [PubMed]
- Cunniffe, S.; O’Neill, P.; Greenberg, M.M.; Lomax, M.E. Reduced repair capacity of a DNA clustered damage site comprised of 8-oxo-7,8-dihydro-2’-deoxyguanosine and 2-deoxyribonolactone results in an increased mutagenic potential of these lesions. Mutat. Res. 2014, 762, 32–39. [Google Scholar] [PubMed]
- Crespan, E.; Pasi, E.; Imoto, S.; Hübscher, U.; Greenberg, M.M.; Maga, G. Human DNA polymerase β, but not λ, can bypass a 2-deoxyribonolactone lesion together with proliferating cell nuclear antigen. ACS Chem. Biol. 2013, 8, 336–344. [Google Scholar] [PubMed]
- Quiñones, J.L.; Thapar, U.; Yu, K.; Fang, Q.; Sobol, R.W.; Demple, B. Enzyme mechanism-based, oxidative DNA–protein cross-links formed with DNA polymerase β in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 8602–8607. [Google Scholar] [PubMed]
- Quiñones, J.L.; Demple, B. When DNA repair goes wrong: BER-generated DNA-protein crosslinks to oxidative lesions. DNA Repair 2016, 44, 103–109. [Google Scholar] [PubMed]
- Stevens, A.J.; Guan, L.; Bebenek, K.; Kunkel, T.A.; Greenberg, M.M. DNA polymerase λ inactivation by oxidized abasic sites. Biochemistry 2013, 52, 975–983. [Google Scholar] [PubMed]
- Blanca, G.; Villani, G.; Shevelev, I.; Ramadan, K.; Spadari, S.; Hübscher, U.; Maga, G. Human DNA polymerases λ and β show different efficiencies of translesion DNA synthesis past abasic sites and alternative mechanisms for frameshift generation. Biochemistry 2004, 43, 11605–11615. [Google Scholar] [PubMed]
- DeMott, M.S.; Beyret, E.; Wong, D.; Bales, B.C.; Hwang, J.-T.; Greenberg, M.M.; Demple, B. Covalent trapping of human DNA polymerase β by the oxidative DNA lesion 2-deoxyribonolactone. J. Biol. Chem. 2002, 277, 7637–7640. [Google Scholar] [PubMed]
- Arian, D.; Hedayati, M.; Zhou, H.; Bilis, Z.; Chen, K.; DeWeese, T.L.; Greenberg, M.M. Irreversible inhibition of DNA polymerase β by small molecule mimics of a DNA lesion. J. Am. Chem. Soc. 2014, 136, 3176–3183. [Google Scholar] [PubMed]
- Liu, L.; Rodriguez-Belmonte, E.M.; Mazloum, N.; Xie, B.; Lee, M.Y. Identification of a novel protein, pdip38, that interacts with the p50 subunit of DNA polymerase δ and proliferating cell nuclear antigen. J. Biol. Chem. 2003, 278, 10041–10047. [Google Scholar] [PubMed]
- Tissier, A.; Janel-Bintz, R.; Coulon, S.; Klaile, E.; Kannouche, P.; Fuchs, R.P.; Cordonnier, A.M. Crosstalk between replicative and translesional DNA polymerases: Pdip38 interacts directly with Polη. DNA Repair 2010, 9, 922–928. [Google Scholar] [PubMed]
- Maga, G.; Crespan, E.; Markkanen, E.; Imhof, R.; Furrer, A.; Villani, G.; Hübscher, U.; van Loon, B. DNA polymerase δ-interacting protein 2 is a processivity factor for DNA polymerase λ during 8-oxo-7,8-dihydroguanine bypass. Proc. Natl. Acad. Sci. USA 2013, 110, 18850–18855. [Google Scholar] [PubMed]
- McElhinny, S.A.N.; Kumar, D.; Clark, A.B.; Watt, D.L.; Watts, B.E.; Lundström, E.-B.; Johansson, E.; Chabes, A.; Kunkel, T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010, 6, 774–781. [Google Scholar]
- Yao, N.Y.; Schroeder, J.W.; Yurieva, O.; Simmons, L.A.; O’Donnell, M.E. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc. Natl. Acad. Sci. USA 2013, 110, 12942–12947. [Google Scholar] [PubMed]
- Clausen, A.R.; Murray, M.S.; Passer, A.R.; Pedersen, L.C.; Kunkel, T.A. Structure–function analysis of ribonucleotide bypass by b family DNA replicases. Proc. Natl. Acad. Sci. USA 2013, 110, 16802–16807. [Google Scholar] [PubMed]
- Reijns, M.A.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 2012, 149, 1008–1022. [Google Scholar] [PubMed]
- Brown, J.A.; Suo, Z. Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry 2011, 50, 1135–1142. [Google Scholar] [PubMed]
- Nick McElhinny, S.A.; Ramsden, D.A. Polymerase mu is a DNA-directed DNA/RNA polymerase. Mol. Cell. Biol. 2003, 23, 2309–2315. [Google Scholar] [PubMed]
- Martin, M.J.; Garcia-Ortiz, M.V.; Esteban, V.; Blanco, L. Ribonucleotides and manganese ions improve non-homologous end joining by human polµ. Nucl. Acids Res. 2013, 41, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- Crespan, E.; Furrer, A.; Rösinger, M.; Bertoletti, F.; Mentegari, E.; Chiapparini, G.; Imhof, R.; Ziegler, N.; Sturla, S.J.; Hübscher, U.; et al. Impact of ribonucleotide incorporation by DNA polymerases β and λ on oxidative base excision repair. Nat. Commun. 2016, 7, 10805. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, R.A.; Moon, A.F.; Kunkel, T.A.; Pedersen, L.C.; Bebenek, K. The catalytic cycle for ribonucleotide incorporation by human DNA Pol λ. Nucl. Acids Res. 2012, 40, 7518–7527. [Google Scholar] [CrossRef]
- Cilli, P.; Minoprio, A.; Bossa, C.; Bignami, M.; Mazzei, F. Formation and repair of mismatches containing ribonucleotides and oxidized bases at repeated DNA sequences. J. Biol. Chem. 2015, 290, 26259–26269. [Google Scholar] [CrossRef] [PubMed]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair 2014, 17, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Crespan, E.; Czabany, T.; Maga, G.; Hübscher, U. Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases λ and β on normal and repetitive DNA sequences. Nucl. Acids Res. 2012, 40, 5577–5590. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.; McCulloch, S.D.; Macpherson, P.; Maga, G.; Yamada, M.; Nohmi, T.; Minoprio, A.; Mazzei, F.; Kunkel, T.A.; Karran, P.; et al. Replication of 2-hydroxyadenine-containing DNA and recognition by human mutsalpha. DNA Repair 2007, 6, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.J.; Brown, G.W. Regulation of chromosome replication. Annu. Rev. Biochem. 2000, 69, 829–880. [Google Scholar] [CrossRef] [PubMed]
- Albertella, M.R.; Lau, A.; O’Connor, M.J. The overexpression of specialized DNA polymerases in cancer. DNA Repair 2005, 4, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Capp, J.-P.; Boudsocq, F.; Bergoglio, V.; Trouche, D.; Cazaux, C.; Blanco, L.; Hoffmann, J.-S.; Canitrot, Y. The R438W polymorphism of human DNA polymerase λ triggers cellular sensitivity to camptothecin by compromising the homologous recombination repair pathway. Carcinogenesis 2010, 31, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Terrados, G.; Capp, J.-P.; Canitrot, Y.; García-Díaz, M.; Bebenek, K.; Kirchhoff, T.; Villanueva, A.; Boudsocq, F.; Bergoglio, V.; Cazaux, C.; et al. Characterization of a natural mutator variant of human DNA polymerase λ which promotes chromosomal instability by compromising NHEJ. PLoS ONE 2009, 4, e7290. [Google Scholar] [CrossRef]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, A.; Ouchida, M.; Miyazaki, K.; Hori, K.; Mukai, T. Functional mutation of DNA polymerase β found in human gastric cancer--inability of the base excision repair in vitro. Mutat. Res. 1999, 435, 121–128. [Google Scholar] [CrossRef]
- Dobashi, Y.; Shuin, T.; Tsuruga, H.; Uemura, H.; Torigoe, S.; Kubota, Y. DNA polymerase β gene mutation in human prostate cancer. Cancer Res. 1994, 54, 2827–2829. [Google Scholar] [PubMed]
- Starcevic, D.; Dalal, S.; Sweasy, J.B. Is there a link between DNA polymerase β and cancer? Cell Cycle 2004, 3, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Donigan, K.A.; Sun, K.W.; Nemec, A.A.; Murphy, D.L.; Cong, X.; Northrup, V.; Zelterman, D.; Sweasy, J.B. Human polb gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 2012, 287, 23830–23839. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Maitra, M.; Starcevic, D.; Li, S.X.; Sweasy, J.B. A DNA polymerase β mutant from colon cancer cells induces mutations. Proc. Natl. Acad. Sci. USA 2004, 101, 6074–6079. [Google Scholar] [CrossRef] [PubMed]
- Nemec, A.A.; Murphy, D.L.; Donigan, K.A.; Sweasy, J.B. The s229l colon tumor-associated variant of DNA polymerase β induces cellular transformation as a result of decreased polymerization efficiency. J. Biol. Chem. 2014, 289, 13708–13716. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Pan, F.; Cao, Y.; Han, Y.; Zhao, J.; Sun, H.; Zhou, X.; Wu, X.; He, L.; Hu, Z.; et al. R152c DNA pol beta mutation impairs base excision repair and induces cellular transformation. Oncotarget 2016, 7, 6902–6915. [Google Scholar] [PubMed]
- Tan, X.; Wang, H.; Luo, G.; Ren, S.; Li, W.; Cui, J.; Gill, H.S.; Fu, S.W.; Lu, Y. Clinical significance of a point mutation in DNA polymerase beta (polb) gene in gastric cancer. Int. J. Biol. Sci. 2015, 11, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S.; Hile, S.; Eckert, K.A.; Sun, K.-W.; Starcevic, D.; Sweasy, J.B. Prostate-cancer-associated i260m variant of DNA polymerase β is a sequence-specific mutator. Biochemistry 2005, 44, 15664–15673. [Google Scholar] [CrossRef] [PubMed]
- Yamtich, J.; Nemec, A.A.; Keh, A.; Sweasy, J.B. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012, 8, e1003052. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zang, W.; Du, Y.; Chen, X.; Zhao, G. The k167i variant of DNA polymerase β that is found in esophageal carcinoma patients impairs polymerase activity and BER. Sci. Rep. 2015, 5, 15986. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Menezes, M.R.; Senejani, A.; Sweasy, J.B. Cellular roles of DNA polymerase β. Yale J. Biol. Med. 2013, 86, 463–469. [Google Scholar] [PubMed]
- Louat, T.; Servant, L.; Rols, M.P.; Bieth, A.; Teissie, J.; Hoffmann, J.S.; Cazaux, C. Antitumor activity of 2′,3′-dideoxycytidine nucleotide analog against tumors up-regulating DNA polymerase β. Mol. Pharmacol. 2001, 60, 553–558. [Google Scholar] [PubMed]
- Bergoglio, V.; Frechet, M.; Philippe, M.; Bieth, A.; Mercier, P.; Morello, D.; Lacroix-Tricki, M.; Delsol, G.; Hoffmann, J.S.; Cazaux, C. Evidence of finely tuned expression of DNA polymerase β in vivo using transgenic mice. FEBS Lett. 2004, 566, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Frechet, M.; Canitrot, Y.; Cazaux, C.; Hoffmann, J.S. DNA polymerase β imbalance increases apoptosis and mutagenesis induced by oxidative stress. FEBS Lett. 2001, 505, 229–232. [Google Scholar] [CrossRef]
- Frechet, M.; Canitrot, Y.; Bieth, A.; Dogliotti, E.; Cazaux, C.; Hoffmann, J.S. Deregulated DNA polymerase β strengthens ionizing radiation-induced nucleotidic and chromosomal instabilities. Oncogene 2002, 21, 2320–2327. [Google Scholar] [CrossRef] [PubMed]
- Fotiadou, P.; Henegariu, O.; Sweasy, J.B. DNA polymerase β interacts with TRF2 and induces telomere dysfunction in a murine mammary cell line. Cancer Res. 2004, 64, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Goula, A.-V.; Merienne, K. Abnormal base excision repair at trinucleotide repeats associated with diseases: A tissue-selective mechanism. Genes 2013, 4, 375. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wilson, S.H. DNA base excision repair: A mechanism of trinucleotide repeat expansion. Trends Biochem. Sci. 2012, 37, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Gu, L.; Leffak, M.; Li, G.-M. MutSβ promotes trinucleotide repeat expansion by recruiting DNA polymerase β to nascent (CAG) n or (CTG) n hairpins for error-prone DNA synthesis. Cell Res. 2016, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Miao, Z.H.; Meng, L.H.; Geng, M.Y. Emerging cancer therapeutic opportunities target DNA-repair systems. Trends Pharmacol. Sci. 2006, 27, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, S.; Hickson, I.D. DNA repair inhibition: A selective tumour targeting strategy. Trends Mol. Med. 2005, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Nakatsuru, Y.; Gerson, S.L. Base excision repair as a therapeutic target in colon cancer. Clin. Cancer Res. 2002, 8, 2985–2991. [Google Scholar] [PubMed]
- Canitrot, Y.; Cazaux, C.; Frechet, M.; Bouayadi, K.; Lesca, C.; Salles, B.; Hoffmann, J.S. Overexpression of DNA polymerase β in cell results in a mutator phenotype and a decreased sensitivity to anticancer drugs. Proc. Natl. Acad. Sci. USA 1998, 95, 12586–12590. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Rechkunova, N.I.; Dezhurov, S.V.; Khodyreva, S.N.; Favre, A.; Blanco, L.; Lavrik, O.I. Comparison of functional properties of mammalian DNA polymerase λ and DNA polymerase β in reactions of DNA synthesis related to DNA repair. Biochim. Biophys. Acta 2005, 1751, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Fiala, K.A.; Abdel-Gawad, W.; Suo, Z. Pre-steady-state kinetic studies of the fidelity and mechanism of polymerization catalyzed by truncated human DNA polymerase λ. Biochemistry 2004, 43, 6751–6762. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Tanaka, N.; Yagi, H.; Kurosawa, T.; Onoue, M.; Seto, H.; Horie, T.; Aoyagi, N.; Yamaoka, M.; Matsukage, A.; et al. Fatty acids selectively inhibit eukaryotic DNA polymerase activities in vitro. Biochim. Biophys. Acta 1996, 1308, 256–262. [Google Scholar] [CrossRef]
- Ono, K.; Nakane, H.; Fukushima, M.; Chermann, J.C.; Barre-Sinoussi, F. Differential inhibitory effects of various flavonoids on the activities of reverse transcriptase and cellular DNA and RNA polymerases. Eur. J. Biochem. 1990, 190, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.H.; Wang, L.K.; Sucheck, S.J.; Snow, A.M.; Hecht, S.M. Inhibitors of DNA polymerase β from schoepfia californica. Chem. Commun. 1998, 2769–2770. [Google Scholar] [CrossRef]
- Tanaka, N.; Kitamura, A.; Mizushina, Y.; Sugawara, F.; Sakaguchi, K. Fomitellic acids, triterpenoid inhibitors of eukaryotic DNA polymerases from a basidiomycete, Fomitella fraxinea. J. Nat. Prod. 1998, 61, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Z.; Sun Di-An, A.; Starck, S.R.; Hecht, S.M.; Cerny, R.L.; Engen, J.R. Chrysochlamic acid, a new diterpenoid-substituted quinol from chrysochlamys ulei that inhibits DNA polymerase β. J. Chem. Soc. Perkin Trans. 1 1999, 24, 1147–1149. [Google Scholar] [CrossRef]
- Sun, D.A.; Deng, J.Z.; Starck, S.R.; Hecht, S.M. Mispyric acid, a new monocyclic triterpenoid with a novel skeleton from Mischocarpus pyriformis that inhibits DNA polymerase β. J. Am. Chem. Soc. 1999, 121, 6120–6124. [Google Scholar] [CrossRef]
- Sun, D.A.; Starck, S.R.; Locke, E.P.; Hecht, S.M. DNA polymerase β inhibitors from Sandoricum koetjape. J. Nat. Prod. 1999, 62, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Z.; Starck, S.R.; Hecht, S.M. DNA polymerase β inhibitors from Baeckea gunniana. J. Nat. Prod. 1999, 62, 1624–1626. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Z.; Starck, S.R.; Hecht, S.M.; Ijames, C.F.; Hemling, M.E. Harbinatic acid, a novel and potent DNA polymerase β inhibitor from Hardwickia binata. J. Nat. Prod. 1999, 62, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Starck, S.R.; Hecht, S.M. DNA polymerase β inhibitors from Tetracera boiviniana. J. Nat. Prod. 1999, 62, 1660–1663. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Z.; Starck, S.R.; Hecht, S.M. Pentacyclic triterpenoids from freziera sp. that inhibit DNA polymerase β. Bioorg. Med. Chem. 2000, 8, 247–250. [Google Scholar] [CrossRef]
- Deng, J.Z.; Starck, S.R.; Sun, D.A.; Sabat, M.; Hecht, S.M. A new 7,8-euphadien-type triterpenoid from Brackenridgea nitida and Bleasdalea bleasdalei that inhibits DNA polymerase β. J. Nat. Prod. 2000, 63, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.M. Inhibitors of the lyase activity of DNA polymerase β. Pharm. Biol. 2003, 41, 68–77. [Google Scholar] [CrossRef]
- Prakash Chaturvedula, V.S.; Gao, Z.; Hecht, S.M.; Jones, S.H.; Kingston, D.G. A new acylated oleanane triterpenoid from Couepia polyandra that inhibits the lyase activity of DNA polymerase β. J. Nat. Prod. 2003, 66, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Gao, Z.; Feng, X.; Jones, S.H.; Hecht, S.M. Plant sterols as selective DNA polymerase β lyase inhibitors and potentiators of Bleomycin cytotoxicity. Bioorg. Med. Chem. 2004, 12, 4253–4258. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Gao, Z.; Feng, X.; Hecht, S.M. Biscoumarin derivatives from edgeworthia gardneri that inhibit the lyase activity of DNA polymerase β. J. Nat. Prod. 2004, 67, 1608–1610. [Google Scholar] [CrossRef] [PubMed]
- Prakash Chaturvedula, V.S.; Hecht, S.M.; Gao, Z.; Jones, S.H.; Feng, X.; Kingston, D.G. New neolignans that inhibit DNA polymerase β lyase. J. Nat. Prod. 2004, 67, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedula, V.S.; Zhou, B.N.; Gao, Z.; Thomas, S.J.; Hecht, S.M.; Kingston, D.G. New lupane triterpenoids from Solidago canadensis that inhibit the lyase activity of DNA polymerase β. Bioorg. Med. Chem. 2004, 12, 6271–6275. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Gao, Z.; Li, S.; Jones, S.H.; Hecht, S.M. DNA polymerase β lyase inhibitors from Maytenus putterlickoides. J. Nat. Prod. 2004, 67, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Murate, T.; Izuta, S.; Takemura, M.; Furuta, K.; Kobayashi, J.; Kamikawa, T.; Nimura, Y.; Yoshida, S. Sulfated glycoglycerolipid from Archaebacterium inhibits eukaryotic DNA polymerase alpha, β and retroviral reverse transcriptase and affects methyl methanesulfonate cytotoxicity. Int. J. Cancer 1998, 76, 512–518. [Google Scholar] [CrossRef]
- Mizushina, Y.; Watanabe, I.; Ohta, K.; Takemura, M.; Sahara, H.; Takahashi, N.; Gasa, S.; Sugawara, F.; Matsukage, A.; Yoshida, S.; et al. Studies on inhibitors of mammalian DNA polymerase α and β: Sulfolipids from a pteridophyte, Athyrium niponicum. Biochem. Pharmacol. 1998, 55, 537–541. [Google Scholar] [CrossRef]
- Gao, Z.; Maloney, D.J.; Dedkova, L.M.; Hecht, S.M. Inhibitors of DNA polymerase β: Activity and mechanism. Bioorg. Med. Chem. 2008, 16, 4331–4340. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.J.; Deng, J.Z.; Starck, S.R.; Gao, Z.; Hecht, S.M. (+)-myristinin A, a naturally occurring DNA polymerase β inhibitor and potent DNA-damaging agent. J. Am. Chem. Soc. 2005, 127, 4140–4141. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Takahashi, N.; Ogawa, A.; Tsurugaya, K.; Koshino, H.; Takemura, M.; Yoshida, S.; Matsukage, A.; Sugawara, F.; Sakaguchi, K. The cyanogenic glucoside, prunasin (D-mandelonitrile-β-D-glucoside), is a novel inhibitor of DNA polymerase β. J. Biochem. 1999, 126, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.S.; Banerjee, S.; Aneja, R.; Sarkar, F.H.; Ostrov, D.A.; Narayan, S. DNA polymerase β as a novel target for chemotherapeutic intervention of colorectal cancer. PLoS ONE 2011, 6, e16691. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Prasad, R.; Hou, E.; Wilson, S.H. Protection against methylation-induced cytotoxicity by DNA polymerase β-dependent long patch base excision repair. J. Biol. Chem. 2000, 275, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Taverna, P.; Liu, L.; Hwang, H.S.; Hanson, A.J.; Kinsella, T.J.; Gerson, S.L. Methoxyamine potentiates DNA single strand breaks and double strand breaks induced by temozolomide in colon cancer cells. Mutat. Res. 2001, 485, 269–281. [Google Scholar] [CrossRef]
- Fishel, M.L.; He, Y.; Smith, M.L.; Kelley, M.R. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin. Cancer Res. 2007, 13, 260–267. [Google Scholar] [CrossRef]
- Tang, J.B.; Svilar, D.; Trivedi, R.N.; Wang, X.H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase β modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-oncology 2011, 13, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, T.; Brockmann, A.; Pott, M.; Hantusch, A.; Brunner, T.; Marx, A. Expanding the scope of human DNA polymerase λ and β inhibitors. ACS Chem. Biol. 2014, 9, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Copani, A.; Sortino, M.A.; Caricasole, A.; Chiechio, S.; Chisari, M.; Battaglia, G.; Giuffrida-Stella, A.M.; Vancheri, C.; Nicoletti, F. Erratic expression of DNA polymerases by β-amyloid causes neuronal death. FASEB J. 2002, 16, 2006–2008. [Google Scholar] [CrossRef] [PubMed]
- Copani, A.; Hoozemans, J.J.; Caraci, F.; Calafiore, M.; Van Haastert, E.S.; Veerhuis, R.; Rozemuller, A.J.; Aronica, E.; Sortino, M.A.; Nicoletti, F. DNA polymerase-β is expressed early in neurons of alzheimer’s disease brain and is loaded into DNA replication forks in neurons challenged with β-amyloid. J. Neurosci. 2006, 26, 10949–10957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cao, X.; Xiong, N.; Wang, H.; Huang, J.; Sun, S.; Liang, Z.; Wang, T. DNA polymerase-β is required for 1-methyl-4-phenylpyridinium-induced apoptotic death in neurons. Apoptosis 2010, 15, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Merlo, S.; Basile, L.; Giuffrida, M.L.; Sortino, M.A.; Guccione, S.; Copani, A. Identification of 5-methoxyflavone as a novel DNA polymerase-β inhibitor and neuroprotective agent against β-amyloid toxicity. J. Nat. Prod. 2015, 78, 2704–2711. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Kamisuki, S.; Kasai, N.; Ishidoh, T.; Shimazaki, N.; Takemura, M.; Asahara, H.; Linn, S.; Yoshida, S.; Koiwai, O.; et al. Petasiphenol: A DNA polymerase λ inhibitor. Biochemistry 2002, 41, 14463–14471. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Hirota, M.; Murakami, C.; Ishidoh, T.; Kamisuki, S.; Shimazaki, N.; Takemura, M.; Perpelescu, M.; Suzuki, M.; Yoshida, H.; et al. Some anti-chronic inflammatory compounds are DNA polymerase λ-specific inhibitors. Biochem. Pharmacol. 2003, 66, 1935–1944. [Google Scholar] [CrossRef]
- Mizushina, Y.; Saito, A.; Tanaka, A.; Nakajima, N.; Kuriyama, I.; Takemura, M.; Takeuchi, T.; Sugawara, F.; Yoshida, H. Structural analysis of catechin derivatives as mammalian DNA polymerase inhibitors. Biochem. Biophys. Res. Commun. 2005, 333, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Kamisuki, S.; Ishimaru, C.; Onoda, K.; Kuriyama, I.; Ida, N.; Sugawara, F.; Yoshida, H.; Mizushina, Y. Nodulisporol and nodulisporone, novel specific inhibitors of human DNA polymerase λ from a fungus, Nodulisporium sp. Bioorg. Med. Chem. 2007, 15, 3109–3114. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Kuriyama, I.; Yoshida, H. Inhibition of DNA polymerase λ and associated inflammatory activities of extracts from steamed germinated soybeans. Food Funct. 2014, 5, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, G.A.; Savio, M.; Forti, L.; Shevelev, I.; Ramadan, K.; Stivala, L.A.; Vannini, V.; Hubscher, U.; Spadari, S.; Maga, G. Inhibition of mammalian DNA polymerases by resveratrol: Mechanism and structural determinants. Biochem. J. 2005, 389, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, T.; Bareth, B.; Immel, T.A.; Huhn, T.; Mayer, T.U.; Marx, A. Small molecule inhibitors of human DNA polymerase λ. ACS Chem. Biol. 2011, 6, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Kamisuki, S.; Kasai, N.; Shimazaki, N.; Takemura, M.; Asahara, H.; Linn, S.; Yoshida, S.; Matsukage, A.; Koiwai, O.; et al. A plant phytotoxin, solanapyrone A, is an inhibitor of DNA polymerase β and λ. J. Biol. Chem. 2002, 277, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Nishida, M.; Kuramochi, K.; Sugawara, F.; Yoshida, H.; Mizushina, Y. Novel azaphilones, kasanosins A and B, which are specific inhibitors of eukaryotic DNA polymerases β and λ from talaromyces sp. Bioorg. Med. Chem. 2008, 16, 4594–4599. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Hada, T.; Kuramochi, K.; Yoshida, H.; Yonezawa, Y.; Kuriyama, I.; Sugawara, F.; Yoshida, H.; Mizushina, Y. Diallyl sulfides: Selective inhibitors of family X DNA polymerases from garlic (Allium sativum L.). Food Chem. 2008, 108, 551–560. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Polymerase β Variant | Human Tumor Type |
|---|---|
| K289M [55] | Colorectal cancer |
| E288K [54] | |
| S229L [56] | |
| R152C [57] | |
| E295K | Gastric cancer |
| G231D | |
| L22P | |
| Y265C | |
| D160N [51] | |
| T889C [58] | |
| I260M [59] | Prostate cancer |
| P242R [60] | Evidence of chromosomal aberrations in human mammary cells |
| K167I [61] | Esophageal cancer |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mentegari, E.; Kissova, M.; Bavagnoli, L.; Maga, G.; Crespan, E. DNA Polymerases λ and β: The Double-Edged Swords of DNA Repair. Genes 2016, 7, 57. https://doi.org/10.3390/genes7090057
Mentegari E, Kissova M, Bavagnoli L, Maga G, Crespan E. DNA Polymerases λ and β: The Double-Edged Swords of DNA Repair. Genes. 2016; 7(9):57. https://doi.org/10.3390/genes7090057
Chicago/Turabian StyleMentegari, Elisa, Miroslava Kissova, Laura Bavagnoli, Giovanni Maga, and Emmanuele Crespan. 2016. "DNA Polymerases λ and β: The Double-Edged Swords of DNA Repair" Genes 7, no. 9: 57. https://doi.org/10.3390/genes7090057
APA StyleMentegari, E., Kissova, M., Bavagnoli, L., Maga, G., & Crespan, E. (2016). DNA Polymerases λ and β: The Double-Edged Swords of DNA Repair. Genes, 7(9), 57. https://doi.org/10.3390/genes7090057