
Integration of Genome and Epigenetic Testing in the Diagnostic Evaluation of Developmental Delay: Differentiating Börjeson–Forssman–Lehmann (BFLS) and White–Kernohan (WHIKERS) Syndromes

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing (GS) and Analytic Pipeline
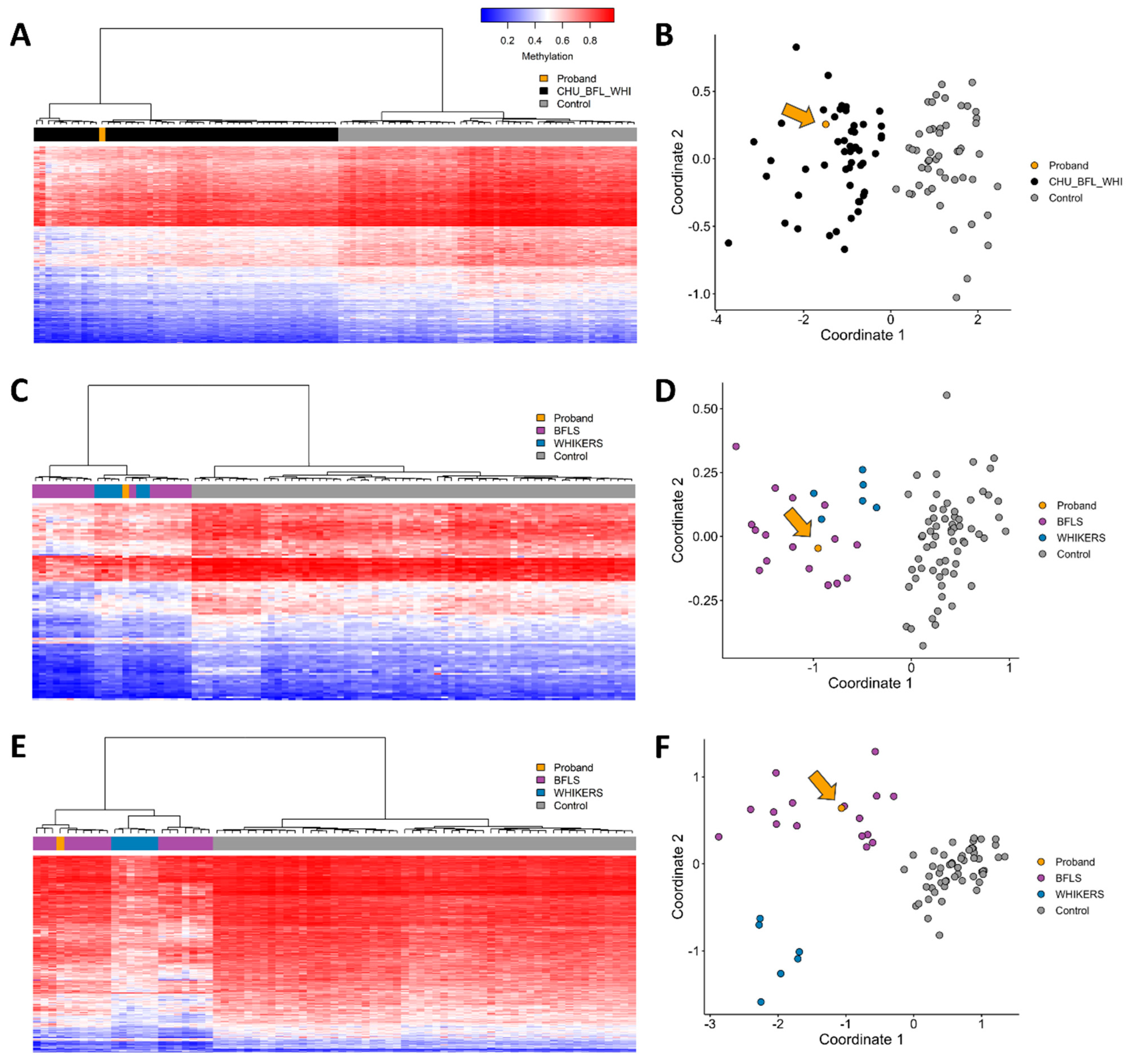
2.2. DNA Methylation Episignature Analysis
3. Results
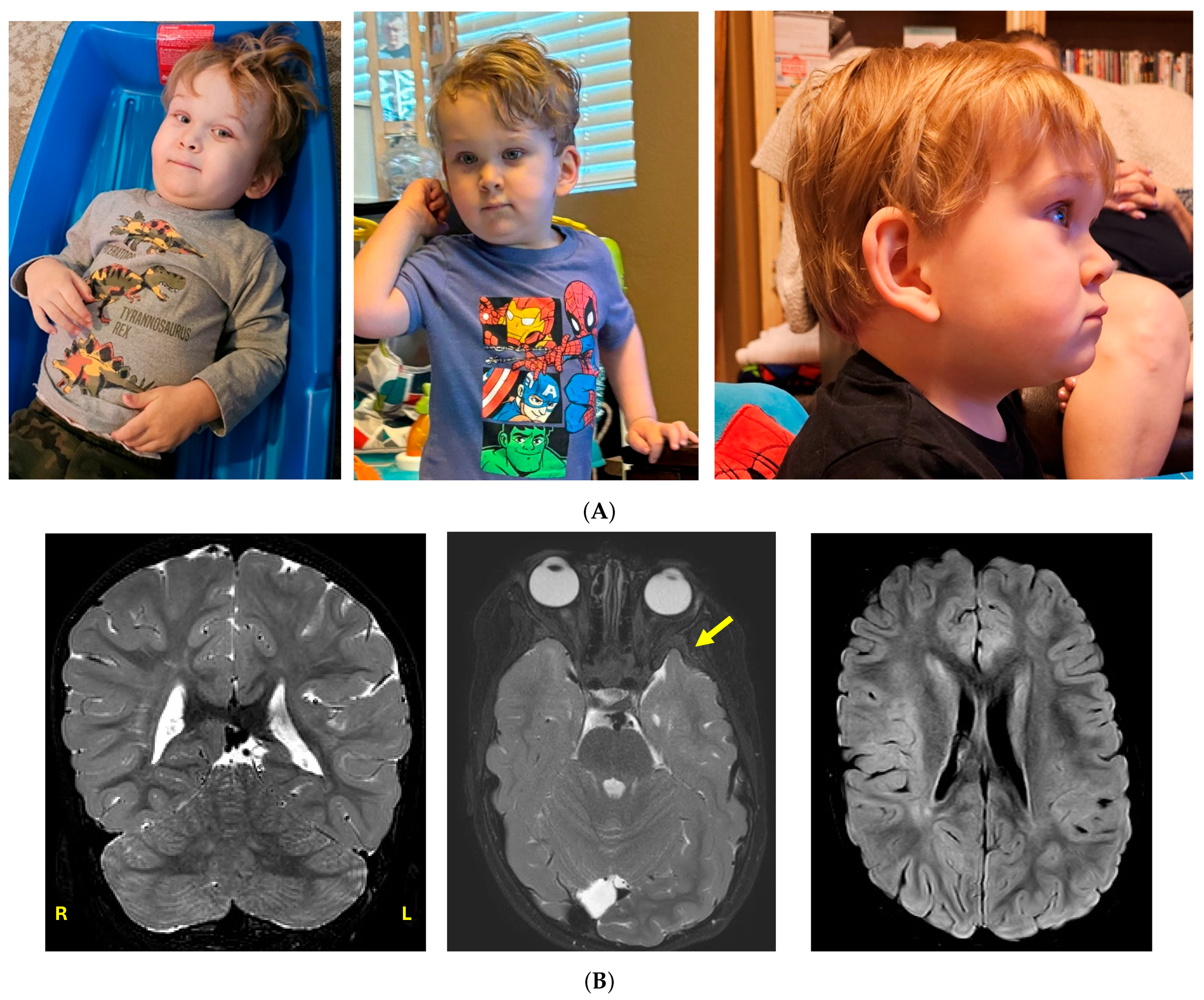
3.1. Clinical Case History
3.2. Genomic Testing
- (1)
- A de novo, likely pathogenic heterozygous variant in SPAST (c.1413+3_1413+6del, NM_014946.3);
- (2)
- A de novo, variant of uncertain significance in DDB1 (c.344 C>T, NM_001923.4, p.P115L);
- (3)
- A maternally inherited, hemizygous variant of uncertain significance in PHF6 (c.763_765del, NM_032458.2, p.T255del). (See Table 1).

{kind=link}
{kind=link}
| Gene Name | Variant | Inheritance | Zygosity | ClinVar ID | ACMG Classification | Population Frequency * | Classification | Clinical Significance |
|---|---|---|---|---|---|---|---|---|
| PHF6 | c.763_765del, NM_032458.2, p.T255del | X-linked Maternal | Hemizygous | 438300 | VUS | Absent | CADD 20.6 | Identified in one other individual with ID and dysmorphic facial features [21]. Clinical overlap in the proband includes intellectual disability, undescended testes, and large ears. |
| DDB1 | c.344 C>T, NM_001923.4, p.P115L | Autosomal Dominant de novo | Heterozygous | - | VUS | Absent | CADD 24.6, REVEL 0.35, BayesDel score of 0.13, and an AlphaMissense score of 0.581 | Clinical overlap in the proband includes developmental delay, large forehead, large ears, differences in his eyebrows (sparse eyebrows spaced farther apart), full cheeks, deep set eyes, epicanthal folds, telecanthus, hypoplastic alae nasi, and 2–3 toe syndactyly. |
| SPAST | c.1413+3_1413+6del | Autosomal Dominant de novo | Heterozygous | 536445 | Pathogenic | Absent | SpliceAI 0.99 | Seen in multiple cases of autosomal dominant hereditary spastic paraplegia (HSP) [14,15,16,17,18,19,20]. Proband does not display signs of spastic paraplegia. |
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| VUS | Variant(s) of uncertain significance |
| DD | Developmental delay |
| ID | Intellectual disability |
| BFLS | Börjeson–Forssman–Lehmann syndrome |
| WHIKERS | White–Keronohan syndrome |
| DNAm | DNA methylation |
| HSP | Hereditary spastic paraplegia |
| MMS1 | Methyl methanesulfonate 1 |
| CPSF | Cleavage and polyadenylation specification factor |
| CADD | Combined annotation dependent depletion |
| REVEL | Rare exome variant ensemble learner |
References
- White, S.M.; Bhoj, E.; Nellåker, C.; Lachmeijer, A.M.; Marshall, A.E.; Boycott, K.M.; Li, D.; Smith, W.; Hartley, T.; McBride, A.; et al. A DNA repair disorder caused by de novo monoallelic DDB1 variants is associated with a neurodevelopmental syndrome. Am. J. Hum. Genet. 2021, 108, 749–756. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lower, K.M.; Turner, G.; Kerr, B.A.; Mathews, K.D.; Shaw, M.A.; Gedeon, Á.K.; Schelley, S.; Hoyme, H.E.; White, S.M.; Delatycki, M.B.; et al. Mutations in PHF6 are associated with Börjeson-Forssman-Lehmann syndrome. Nat. Genet. 2002, 32, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443, Erratum in Nature 2021, 597, E3–E4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Börjeson, M.; Forssman, H.; Lehmann, O. An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Medica Scand. 1962, 171, 13–21. [Google Scholar] [CrossRef] [PubMed]
- McRae, H.M.; Leong, M.P.Y.; Bergamasco, M.I.; Garnham, A.L.; Hu, Y.; Corbett, M.A.; Whitehead, L.; El-Saafin, F.; Sheikh, B.N.; Wilcox, S.; et al. Loss of PHF6 causes spontaneous seizures, enlarged brain ventricles and altered transcription in the cortex of a mouse model of the Börjeson-Forssman-Lehmann intellectual disability syndrome. PLoS Genet. 2024, 20, e1011428. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hameed, M.; Siddiqui, F.; Sheikh, F.H.; Khan, M.K.; Admani, B.; Gangishetti, P.K. Börjeson-Forssman-Lehmann Syndrome: Clinical Features and Diagnostic Challenges. Brain Neurorehabil. 2023, 16, e32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vos, N.; Haghshenas, S.; van der Laan, L.; Russel, P.K.M.; Rooney, K.; Levy, M.A.; Relator, R.; Kerkhof, J.; McConkey, H.; Maas, S.M.; et al. The detection of a strong episignature for Chung-Jansen syndrome, partially overlapping with Börjeson-Forssman-Lehmann and White-Kernohan syndromes. Hum. Genet. 2024, 143, 761–773. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jepsen, W.M.; Fazenbaker, A.; Ramsey, K.; Bonfitto, A.; Naymik, M.; Turner, B.; Sloan, J.; Tiwari, N.; Bernes, S.M.; Neilson, D.E.; et al. Duchenne Muscular Dystrophy in Two Half-Brothers Due to Inherited 306 Kb Inverted Insertion of 10p15.1 into Intron 44 of the Dp427m Transcript of the DMD Gene. Int. J. Mol. Sci. 2024, 25, 11922. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; DI France, G.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370, Erratum in Am. J. Hum. Genet. 2021, 108, 1161–1163. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: Genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 23, 1065–1074, Erratum in Genet. Med. 2021, 23, 2228. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Levy, M.A.; McConkey, H.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; Ciolfi, A.; Clarke, A.; et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 2021, 3, 100075. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kerkhof, J.; Rastin, C.; Levy, M.A.; Relator, R.; McConkey, H.; Demain, L.; Dominguez-Garrido, E.; Kaat, L.D.; Houge, S.D.; DuPont, B.R.; et al. Diagnostic utility and reporting recommendations for clinical DNA methylation episignature testing in genetically undiagnosed rare diseases. Genet. Med. 2024, 26, 101075. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fonknechten, N.; Mavel, D.; Byrne, P.; Davoine, C.S.; Cruaud, C.; Bönsch, D.; Samson, D.; Coutinho, P.; Hutchinson, M.; McMonagle, P.; et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum. Mol. Genet. 2000, 9, 637–644, Erratum in Hum. Mol. Genet. 2005, 14, 461. Boentsch, D [corrected to Bönsch, D]. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Scarano, V.; Cricchi, F.; Melone, M.A.B.; Chiriaco, M.; Napolitano, A.; Malandrini, A.; De Michele, G.; Petrozzi, L.; Giraldi, C.; et al. Autosomal dominant hereditary spastic paraplegia: DHPLC-based mutation analysis of SPG4 reveals eleven novel mutations. Hum. Mutat. 2005, 25, 506. [Google Scholar] [CrossRef] [PubMed]
- Vandebona, H.; Kerr, N.P.; Liang, C.; Sue, C.M. SPAST mutations in Australian patients with hereditary spastic paraplegia. Intern. Med. J. 2012, 42, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, G.; Fan, Y.; Matigian, N.; Wali, G.; Bellette, B.; Sutharsan, R.; Raju, J.; Wood, S.A.; Veivers, D.; Sue, C.M.; et al. A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis. Model. Mech. 2013, 6, 489–502, Erratum in Dis. Model. Mech. 2015, 8, 1339. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Elert-Dobkowska, E.; Stepniak, I.; Krysa, W.; Rajkiewicz, M.; Rakowicz, M.; Sobanska, A.; Rudzinska, M.; Wasielewska, A.; Pilch, J.; Kubalska, J.; et al. Molecular spectrum of the SPAST, ATL1 and REEP1 gene mutations associated with the most common hereditary spastic paraplegias in a group of Polish patients. J. Neurol. Sci. 2015, 359, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Seo, J.Y.; Lee, K.W.; Park, H.M. Novel Pathogenic Variant of SPAST (c.1413 + 4A>G) in a Patient with Hereditary Spastic Paraplegia. J. Clin. Neurol. 2019, 15, 120–121. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Parodi, L.; Fenu, S.; Barbier, M.; Banneau, G.; Duyckaerts, C.; du Montcel, S.T.; Monin, M.L.; Said, S.A.; Guegan, J.E.; Tallaksen, C.M.E.; et al. Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain 2018, 141, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.M.; Adam, S.; du Souich, C.; Lehman, A.; Nelson, T.N.; van Karnebeek, C.; Alderman, E.; Armstrong, L.; Aubertin, G.; Blood, K.; et al. Genome-wide sequencing and the clinical diagnosis of genetic disease: The CAUSES study. HGG Adv. 2022, 3, 100108. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Svenson, I.K.; Kloos, M.T.; Gaskell, P.C.; Nance, M.A.; Garbern, J.Y.; Hisanaga, S.I.; Pericak-Vance, M.A.; Ashley-Koch, A.E.; Marchuk, D.A. Intragenic modifiers of hereditary spastic paraplegia due to spastin gene mutations. Neurogenetics 2004, 5, 157–164. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsey, K.; Prakash, S.; Kerkhof, J.; Sadikovic, B.; White, S.; Naymik, M.; Sloan, J.; Bonfitto, A.; Belnap, N.; Sanchez-Castillo, M.; et al. Integration of Genome and Epigenetic Testing in the Diagnostic Evaluation of Developmental Delay: Differentiating Börjeson–Forssman–Lehmann (BFLS) and White–Kernohan (WHIKERS) Syndromes. Genes 2025, 16, 933. https://doi.org/10.3390/genes16080933
Ramsey K, Prakash S, Kerkhof J, Sadikovic B, White S, Naymik M, Sloan J, Bonfitto A, Belnap N, Sanchez-Castillo M, et al. Integration of Genome and Epigenetic Testing in the Diagnostic Evaluation of Developmental Delay: Differentiating Börjeson–Forssman–Lehmann (BFLS) and White–Kernohan (WHIKERS) Syndromes. Genes. 2025; 16(8):933. https://doi.org/10.3390/genes16080933
Chicago/Turabian StyleRamsey, Keri, Supraja Prakash, Jennifer Kerkhof, Bekim Sadikovic, Susan White, Marcus Naymik, Jennifer Sloan, Anna Bonfitto, Newell Belnap, Meredith Sanchez-Castillo, and et al. 2025. "Integration of Genome and Epigenetic Testing in the Diagnostic Evaluation of Developmental Delay: Differentiating Börjeson–Forssman–Lehmann (BFLS) and White–Kernohan (WHIKERS) Syndromes" Genes 16, no. 8: 933. https://doi.org/10.3390/genes16080933
APA StyleRamsey, K., Prakash, S., Kerkhof, J., Sadikovic, B., White, S., Naymik, M., Sloan, J., Bonfitto, A., Belnap, N., Sanchez-Castillo, M., Jepsen, W., Huentelman, M., Bernes, S., Narayanan, V., & Kaur, S. (2025). Integration of Genome and Epigenetic Testing in the Diagnostic Evaluation of Developmental Delay: Differentiating Börjeson–Forssman–Lehmann (BFLS) and White–Kernohan (WHIKERS) Syndromes. Genes, 16(8), 933. https://doi.org/10.3390/genes16080933