

Community Composition and Diversity of β-Glucosidase Genes in Soils by Amplicon Sequence Variant Analysis

Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sampling
2.2. DNA Extraction and PCR Amplification of β-Glucosidase Genes in Soils
2.3. Next Generation Sequencing Analysis of β-Glucosidase Genes in Soils
2.4. Phylogenetic Analysis of Unclassified β-Glucosidase Genes
3. Results
3.1. β-Glucosidase Genes Soil Communities Based upon Phyla
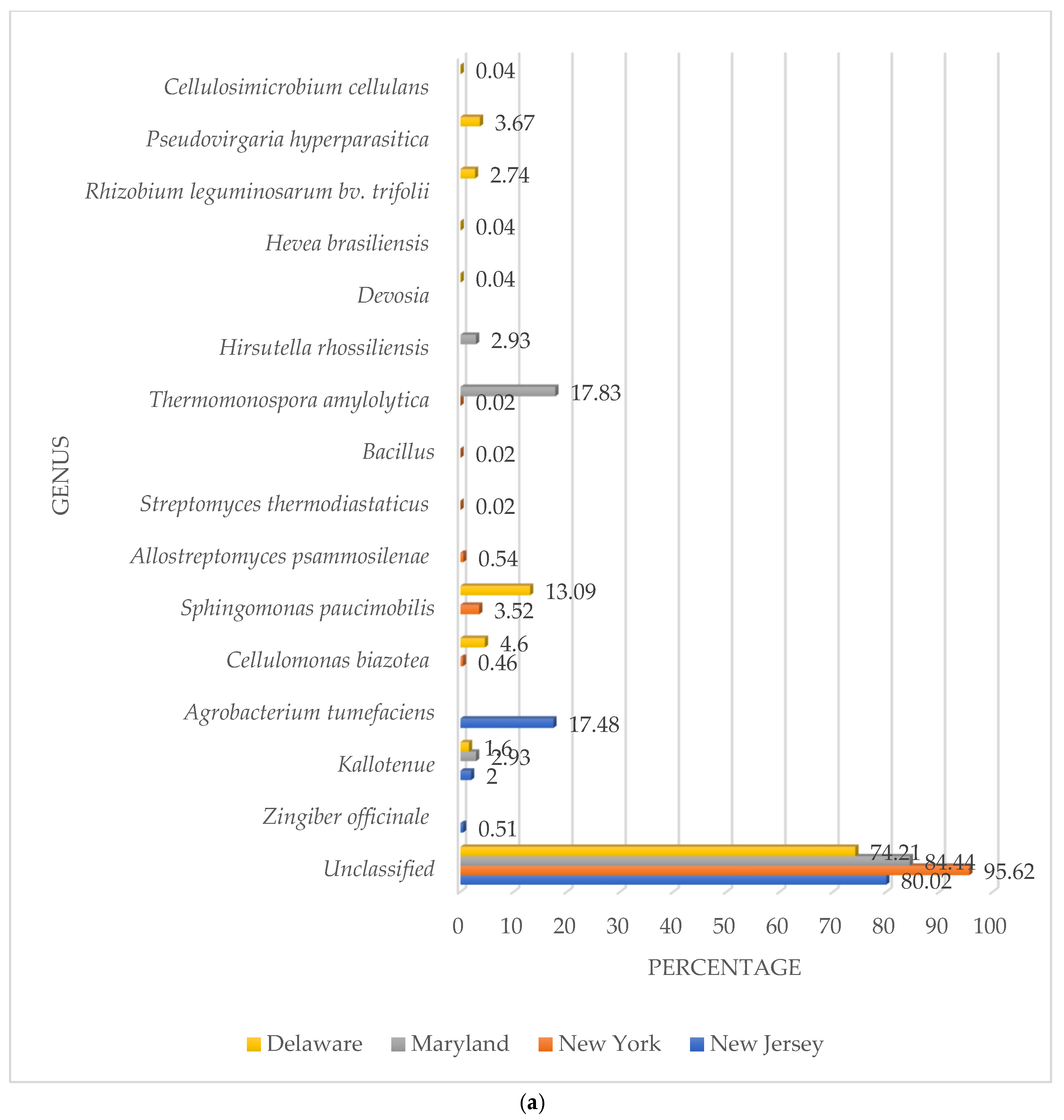
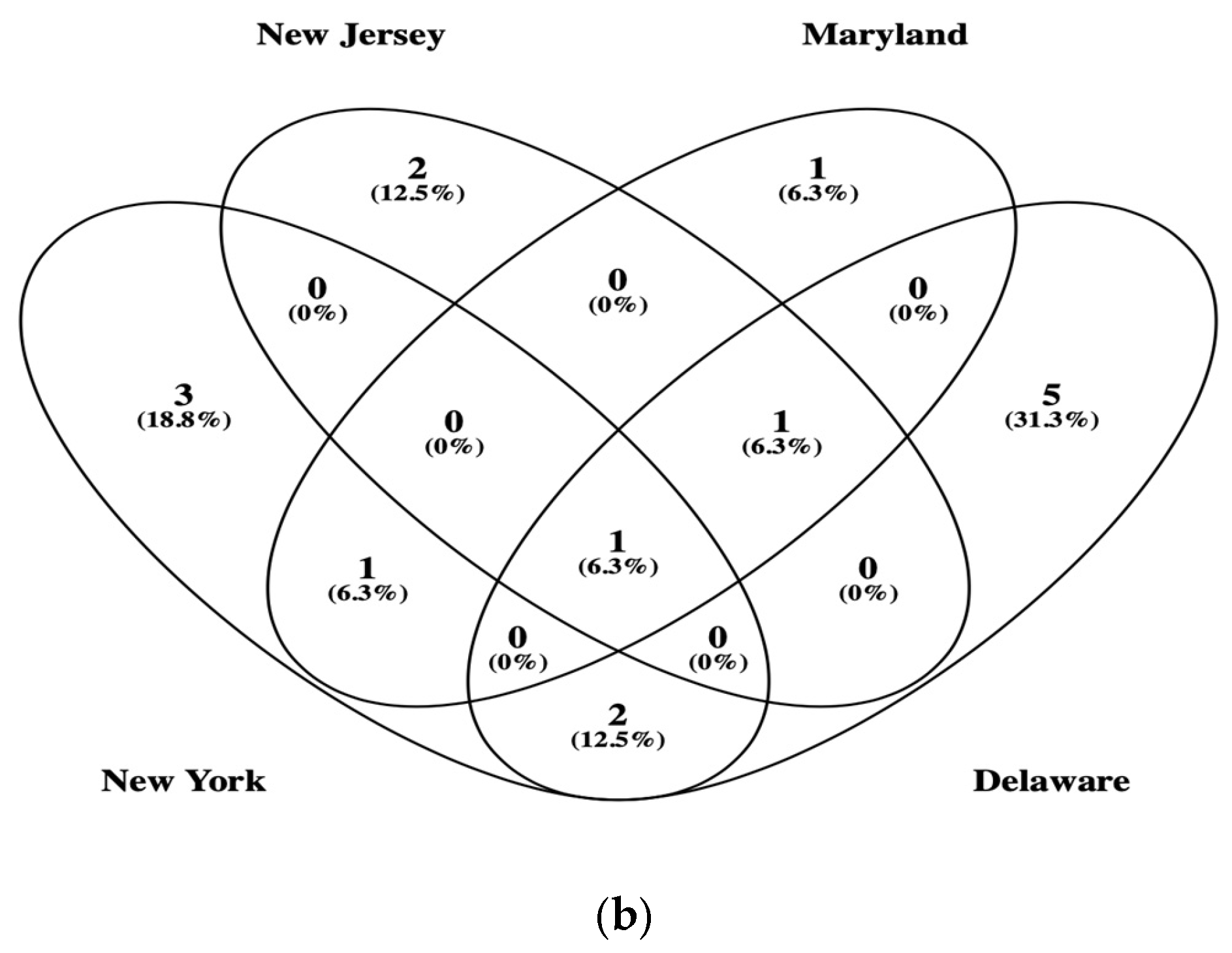
3.2. β-Glucosidase Gene Soil Communities Based upon Bacteria, Fungi, and Plant Genera
3.3. Phylogenetic Analysis of β-Glucosidase Gene Sequences
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Mondejar, R.; Zuhike, D.; Becher, D.; Riedel, K.; Baldrian, P. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 2016, 6, 25279. [Google Scholar] [CrossRef]
- Karimi, B.; Terrat, S.; Dequiedt, S.; Saby, N.P.A.; Horrigue, W.; Lelievre, M.; Nowak, V.; Jolivet, C.; Arrouays, D.; Wincker, P.; et al. Biogeography of soil bacteria and archaea across France. Sci. Adv. 2018, 4, eaat1808. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-Gonzalez, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef]
- Llado, S.; Lopez-Mondejar, R.; Baldrian, P. Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 2017, 81, 10–1128. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Global diversity and geography of soil fungi. Science 2014, 346, 1256688. [Google Scholar] [CrossRef]
- Escalas, A.; Hale, L.; Voordeckers, J.W.; Yang, Y.; Firestone, M.K.; Alvarez-Cohen, L.; Zhou, J. Microbial functional diversity: From concepts to applications. Ecol. Evol. 2019, 9, 12000–12016. [Google Scholar] [CrossRef] [PubMed]
- Cholet, F.; Lisik, A.; Agogue, H.; Ijaz, U.Z.; Pineau, P.; Lachaussee, N.; Smith, C.J. Ecological observations based upon on functional gene sequencing are sensitive to the amplicon method. mSphere 2022, 7, e0032422. [Google Scholar] [CrossRef]
- Aigle, A.; Prosser, J.I.; Gubry-Rangin, C. The application of high-throughput sequencing technology to analysis of amoA phylogeny and environmental niche specialisation of terrestrial bacterial ammonia-oxidizers. Environ. Microbiome 2019, 14, 3. [Google Scholar] [CrossRef]
- Kozjek, K.; Manoharan, L.; Ahren, D.; Hedlund, K. Microbial functional genes influenced by short-term experimental drought across European agricultural fields. Soil Biol. Biochem. 2022, 168, 108650. [Google Scholar] [CrossRef]
- Drazenka, S.; Schmid, M.; Hartmann, A. Diversity of green-like and red-like ribulose-1,5-bisphosphate carboxylase/oxygenase large-subunit genes (cbbL) in differently managed agricultural soils. Appl. Environ. Microbiol. 2005, 71, 175–184. [Google Scholar]
- Poly, F.; Wertz, S.; Brothier, E.; Degrange, V. First exploration of Nitrobacter diversity in soils by a PCR-cloning-sequencing approach targeting functional gene nxrA. FEMS Microbiol. Ecol. 2008, 63, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Rotthauwe, J.H.; Witzel, K.P.; Liesack, W. The ammonia monooxygenase structural gene amoA as a functional marker: Molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microbiol. 1997, 63, 4704–4712. [Google Scholar] [CrossRef]
- Jimenez, L.; Turku, A.; Pincus, L. Genetic diversity and community structure of bacterial glycoside hydrolases family 48 genes in soil and compost. BIOS 2024, 95, 125–133. [Google Scholar] [CrossRef]
- Jimenez, L.; Vazquez, S.; Turku, A.; Pincus, L. PCR detection, cloning, and genetic identification of microbial cellulases in soils. BIOS 2022, 93, 31–48. [Google Scholar] [CrossRef]
- Choi, J.; Bach, E.; Lee, J.; Dooley, S.; Howe, A.; Hofmockel, K.S. Spatial structuring of cellulase gene abundance and activity in soil. Front. Environ. Sci. 2018, 6, 107. [Google Scholar] [CrossRef]
- Hazen, T.C.; Chakraborty, R.; Fleming, J.M.; Gregory, I.R.; Bowman, J.P.; Jimenez, L.; Zhang, D.; Pfiffner, S.M.; Brockman, F.J.; Sayler, G.S. Use of gene probes to assess the impact and effectiveness of aerobic in situ bioremediation of TCE. Arch. Microbiol. 2009, 191, 221–232. [Google Scholar] [CrossRef]
- Pereyra, L.P.; Hiibel, S.R.; Prieto Riquelme, M.V.; Reardon, K.F.; Pruden, A. Detection and quantification of functional genes of cellulose-degrading, fermentative, and sulfate-reducing bacteria and methanogenic archaea. Appl. Environ. Microbiol. 2010, 76, 2192–2202. [Google Scholar] [CrossRef]
- Rettenmaier, R.; Lo, Y.K.; Schmidt, L.; Munk, B.; Lagkouvardos, I.; Neuhaus, K.; Schwarz, W.; Liebl, W.; Zverlov, V. A novel primer mixture for GH48 genes: Quantification and identification of truly cellulolytic bacteria in biogas fermenters. Microorganisms 2020, 8, 1297. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Phylogenetic distribution of potential cellulases in bacteria. Appl. Environ. Microbiol. 2013, 79, 1545–1554. [Google Scholar] [CrossRef]
- Berlemont, R. The supragenic organization of glycoside hydrolase encoding genes reveals distinct strategies for carbohydrate utilization in bacteria. Front. Microbiol. 2023, 14, 1179206. [Google Scholar] [CrossRef]
- Hua, M.; Yu, S.; Ma, Y.; Chen, S.; Li, F. Genetic diversity detection and gene discovery of novel glycoside hydrolase familty 48 from soil environmental genomic DNA. Ann. Microbiol. 2018, 68, 163–174. [Google Scholar] [CrossRef]
- Koeck, D.E.; Pechtl, A.; Zverlov, V.V.; Schwarz, W.H. Genomics of cellulolytic bacteria. Curr. Opin. Biotechnol. 2014, 29, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Moreno, B.; Cañizares, R.; Nuñez, R.; Benitez, E. Genetic diversity of bacterial β-glucosidase-encoding genes as a function of soil management. Biol. Fertil. Soils 2013, 49, 735–745. [Google Scholar] [CrossRef]
- Li, H.; Xu, X.; Chen, H.; Zhing, Y.; Xu, J.; Wang, J.; Lu, X. Molecular analysis of the functional microbial community in composting by PCR-DGGE targeting the genes of the beta-glucosidase. Bioresour. Technol. 2013, 134, 51–58. [Google Scholar] [CrossRef]
- Pathan, S.I.; Zifcakova, L.; Ceccherini, M.T.; Pantani, O.L.; Vetrovsky, T.; Baldrian, P. Seasonal variation and distribution of total and active microbial community of β-glucosidase encoding genes in coniferous forest soil. Soil Biol. Biochem. 2017, 105, 71–80. [Google Scholar] [CrossRef]
- Sacca, M.L.; Caputo, F.; Enrico, C.; Flavio, F. Fungal glucosidase gene and corresponding enzyme activity are positively related to soil organic carbon in unmanaged woody plantations. Soil Ecol. Lett. 2024, 6, 240238. [Google Scholar] [CrossRef]
- Magwaza, B.; Amobonye, A.; Pillai, S. Microbial β-glucosidases: Recent advances and applications. Biochemie 2024, 225, 49–67. [Google Scholar] [CrossRef]
- Barbi, F.; Bragalini, C.; Vallon, L.; Prudent, E.; Dubost, A.; Fraissinet-Tachet, L.; Marmeisse, R.; Luis, P. PCR primers to study the diversity of expressed fungal genes encoding lignocellulolytic enzymes in soils using high-throughput sequencing. PLoS ONE 2014, 9, e116264. [Google Scholar] [CrossRef]
- Tiwari, R.; Kumar, K.; Singh, S.; Nain, L.; Shukla, P. Molecular detection and environmentla-specific diversity of glycosyl hydrolase family 1 β-glucosidase in different habitats. Front. Microbiol. 2016, 7, 1597. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L.; Canal Delgado, I.; Barron, E.; Zapata, S.; Jashari, T.; Choe, T.; Melkonyan, S. Direct PCR detection, cloning, and characterization of bacterial β-glucosidase genes from temperate soils. EC Microbiol. 2015, 2, 261–268. [Google Scholar]
- Zeng, Q.; Mei, T.; Wang, M.; Tan, W. Intensive citrus plantation suppress the microbial profiles of the β-glucosidase gene. Agric. Ecosyst. Environ. 2022, 324, 107687. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, J.; Liu, Q.; Fu, Q.; Hu, H.; Huang, Q. Role of genes encoding microbial carbohydrate-active enzymes in the accumulation and dynamics of organic carbon in subtropical forest soils. Sci. Total Environ. 2024, 918, 170295. [Google Scholar] [CrossRef]
- Yang, X.; Ni, K.; Shi, Y.; Yi, X.; Ji, L.; Wei, S.; Jiang, Y.; Zhang, Y.; Cai, Y.; Ma, Q.; et al. Metagenomics reveals N-induced changes in carbon-degrading genes and microbial communities of tea (Camellia sinensis L.) plantation soil under long-term fertilization. Sci. Total Environ. 2023, 856, 159231. [Google Scholar] [CrossRef]
- Jimenez, L. Bacterial community composition and diversity of soils from different geographical locations in the Northeastern USA. Microbiol. Res. 2025, 16, 47. [Google Scholar] [CrossRef]
- Jimenez, L.; Kulko, M.; Kim, R.; Jashari, T.; Choe, T. 16S rRNA analysis of electrogenic bacterial communities in microbial fuel cells developed from temperate soils. BIOS 2020, 91, 9–20. [Google Scholar] [CrossRef]
- Jimenez, L.; Jashari, T.; Vasquez, J.; Zapata, S.; Bochis, J.; Kulko, M.; Ellman, V.; Gardner, M.; Choe, T. Real-Time PCR detection of Burkholderia cepacia in pharmaceutical products contaminated with low levels of bacterial contamination. PDA J. Pharm. Sci. Technol. 2018, 72, 73–80. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operations taxonomic units in marker-gene analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acid Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Behnke-Borowczyk, J.; Korzeniewicz, R.; Lukoski, A.; Baranowska, M.; Jagiello, R.; Bulaj, B.; Hauke-Kowalska, M.; Szmyt, J.; Behnke, J.M.; Robakowski, P.; et al. Variability of functional groups of rhizosphere fungi of Norway spruce (Picea abies (L.) H. Karst.) in the boreal range: The Wigry national park, Poland. Int. J. Mol. Sci. 2023, 24, 12628. [Google Scholar] [CrossRef]
- Real, R.; Vargas, J.M. The probabilistic basis of Jaccard’s index of similarity. Syst. Biol. 1996, 45, 380–385. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acid Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Berlemont, R.; Allison, S.D.; Weihe, C.; Lu, Y.; Brodie, E.L.; Martiny, J.B.H.; Martiny, A.C. Cellulolytic potential under environmental changes in microbial communities from grassland litter. Front. Microbiol. 2014, 5, 639. [Google Scholar] [CrossRef]
- Fu, Y.; Yin, Z.; Wu, L.; Yin, C. Diversity of cultivable glycosidase-producing microorganisms isolated from the soil of a ginseng field and their ginsenosides hydrolysing activity. Lett. Appl. Microbiol. 2014, 58, 138–144. [Google Scholar] [CrossRef]
- Chan, A.K.N.; Wang, Y.Y.; Ng, K.L.; Fu, Z.; Wong, W.K.R. Cloning and characterization of a novel cellobiase gene, cba3, encoding the first known β-glucosidase of glycosidase hydrolase family 1 of Cellulomonas biazotea. Gene 2012, 493, 52–61. [Google Scholar] [CrossRef]
- Siddique, F.; Lam, E.K.H.; Wong, W.K.R. Syngergistic hydrolysis of filter paper by recombinant cellulase cocktails leveraging a key celllobiase, Cba2, of Cellulomonas biazotea. Front. Bioeng. Biotechnol. 2022, 10, 990984. [Google Scholar] [CrossRef] [PubMed]
- Braun, U.; Crous, P.W.; Groenewald, J.Z.; Scheuer, C. Pseudovirgaria, a fungicolous hyphomycete genus. IMA Fungus 2011, 2, 65–69. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, Y.; Jiang, X.; Shu, C.; Imran Hamid, M.; Hussain, M.; Chen, S.; Xu, J.; Xiang, M.; Liu, X. Population genetics of Hirsutella rhossiliensis, a dominant parasite of cyst nematode juveniles on a continental scale. Appl. Environ. Microbiol. 2016, 82, 6317–6325. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Jiang, Y.; Hur, Y. Genome-wide analysis of glycoside hydrolase family 1 β-glucosidase genes in Brassica rapa and their potential role in pollen development. Int. J. Mol. Sci. 2019, 20, 1663. [Google Scholar] [CrossRef]
- Opassiri, R.; Pomthong, B.; Onkoksooong, T.; Akiyama, T.; Esen, A.; Ketudat Cairns, J.R. Analysis of rice glycosyl hydrolase family 1, expression of Os4bglu12 β-glucosidase. BMC Plant Biol. 2006, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zheng, Y.; Yu, K.; Gao, S.; Zhao, X.; Cao, A.; Han, Q. Technology for distribution and control of Agrobacterium tumefaciens in cherry tree soil. Agriculture 2024, 14, 1857. [Google Scholar] [CrossRef]
- Barton, I.S.; Fuqua, C.; Platt, T.G. Ecological and evolutionary dynamics of a model facultative pathogen: Agrobacterium and crown gall disease of plants. Environ. Microbiol. 2017, 20, 16–29. [Google Scholar] [CrossRef]
- Krimi, Z.; Petit, A.; Mougel, C.; Dessaus, Y.; Nesme, X. Seasonal fluctuations and long-term persistence of populations of Agrobacterium spp. in soils. Appl. Environ. Microbiol. 2002, 68, 3358–3365. [Google Scholar] [CrossRef]
- Wang, C.; Ye, F.; Chang, C.; Liu, X.; Wang, J.; Wang, J.; Yan, X.F.; Fu, Q.; Zhou, J.; Chen, S.; et al. Agrobacteria reprogram virulence gene expression by controlled released of host-conjugated signals. Proc. Natl. Acad. USA 2019, 44, 22331–22340. [Google Scholar] [CrossRef]
- Jiao, J.Y.; Liu, L.; Zhou, E.M.; Wei, D.Q.; Ming, H.; Xian, W.D.; Yuan, C.G.; Zhong, J.M.; Li, W.J. Actinomadura amylolytica sp. nov. and Actinomadura cellulosilytica sp. nov., isolated from geothermally heated soil. Antonie Van Leeuwenhoek 2015, 108, 75–83. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Santana, M.M.; Gomez, E.J.; Delgado, J.A. Soil thermophiles and their extracellular enzymes: A set of capabilites able to provide significant services and risks. Microorganisms 2023, 11, 1650. [Google Scholar] [CrossRef]
- Gomez, E.J.; Delgado, J.A.; Gonzalez, J.M. Persistence of microbial extracellular enzymes in soils under different temperatures and water availabilities. Ecol. Evol. 2020, 10, 10167–10176. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.R.; Sang, P.; Xian, W.D.; Li, X.; Jiao, J.Y.; Liu, L.; Hozzein, W.N.; Xiao, M.; Li, W.J. Expression and characteristics of two glucose-tolerant GH1 β-glucosidases from Actinomadura amylolytica YOM77502 for promoting cellulose degradation. Front. Microbiol. 2018, 9, 3149. [Google Scholar] [CrossRef]
- Cole, J.K.; Gieler, B.A.; Heisler, D.L.; Palisoc, M.M.; Williams, A.J.; Dohnalkova, A.C.; Ming, H.; Yu, T.T.; Dodsworth, J.A.; Li, W.J.; et al. Kallotenue papylolyticum gen. nov., sp. nov., a cellulolytic and filamentous thermophile that represents a novel linage (Kallotenuales ord. nov., Kallotenuaceae fam. nov.) within the class Chlofoflexia. Int. J. Syst. Evol. Microbiol. 2013, 63, 4674–4682. [Google Scholar] [CrossRef]
- Hedlund, B.P.; Murugapiran, S.K.; Huntemann, M.; Clum, A.; Pillay, M.; Palaniappan, K.; Varghese, N.; Mikhailova, N.; Stamatis, D.; Reddy, T.B.K.; et al. High-quality draft genome sequences of Kallotenue papyrolyticum JKG2T reveals broad heterotrophic capacity focused on carbohydrate and amino acid metabolism. Genome Announc. 2015, 3, e01410–e01415. [Google Scholar] [CrossRef]
- Lopez-Lozano, N.E.; Echeverria Molinar, A.; Ortiz Duran, E.A.; Hernandez Rosales, M.; Souza, V. Bacterial diversity and interaction of Agave lechuguilla rhizosphere differ significantly from bulk soil in the oligotrophic basin of Cuatro Cienagas. Front. Plant Sci. 2020, 11, 1028. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, C.; Yang, H. Responses of Cyanobacterial crusts and microbial communities to extreme environments of the stratosphere. Microorganisms 2022, 10, 1252. [Google Scholar] [CrossRef] [PubMed]
- Habib, R.; Do, M.P.; Chen, Y.; Jian, G.; Sivakumar, M. Elucidating biofouling development and succession in membrane distillation using treated effluent. Environ. Res. 2024, 262, 119864. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, B.; Liu, J.; Chen, X.; Li, S.; Su, E.; Gao, L.; Li, H. β-Glucosidase genes differentially expressed during composting. Biotechnol. Biofuels 2020, 13, 174. [Google Scholar] [CrossRef]
- Emre Erkani, M.; El-Halabi, K.; Ryoun Kim, J. Exploring the diversity of glucosidase: Classification, catalytic mechanism, molecular characteristics, kinetic models, and applications. Enzym. Microb. Technol. 2024, 173, 110363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus/Species | Accession Number |
|---|---|
| Agrobacterium tumefaciens | KU512832.1 |
| Cellulomonas biazotea | JF727823.1 |
| Kallotenue | OR724869.1 |
| Sphingomonas paucimobilis | MW804636.1 |
| Thermomonospora amylolytica | MH974517.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez, L. Community Composition and Diversity of β-Glucosidase Genes in Soils by Amplicon Sequence Variant Analysis. Genes 2025, 16, 900. https://doi.org/10.3390/genes16080900
Jimenez L. Community Composition and Diversity of β-Glucosidase Genes in Soils by Amplicon Sequence Variant Analysis. Genes. 2025; 16(8):900. https://doi.org/10.3390/genes16080900
Chicago/Turabian StyleJimenez, Luis. 2025. "Community Composition and Diversity of β-Glucosidase Genes in Soils by Amplicon Sequence Variant Analysis" Genes 16, no. 8: 900. https://doi.org/10.3390/genes16080900
APA StyleJimenez, L. (2025). Community Composition and Diversity of β-Glucosidase Genes in Soils by Amplicon Sequence Variant Analysis. Genes, 16(8), 900. https://doi.org/10.3390/genes16080900