
Global Transcriptome and Weighted Gene Co-Expression Network Analyses of Cold Stress Responses in Chinese Cabbage

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Cold (Low-Temperature) Treatment Conditions
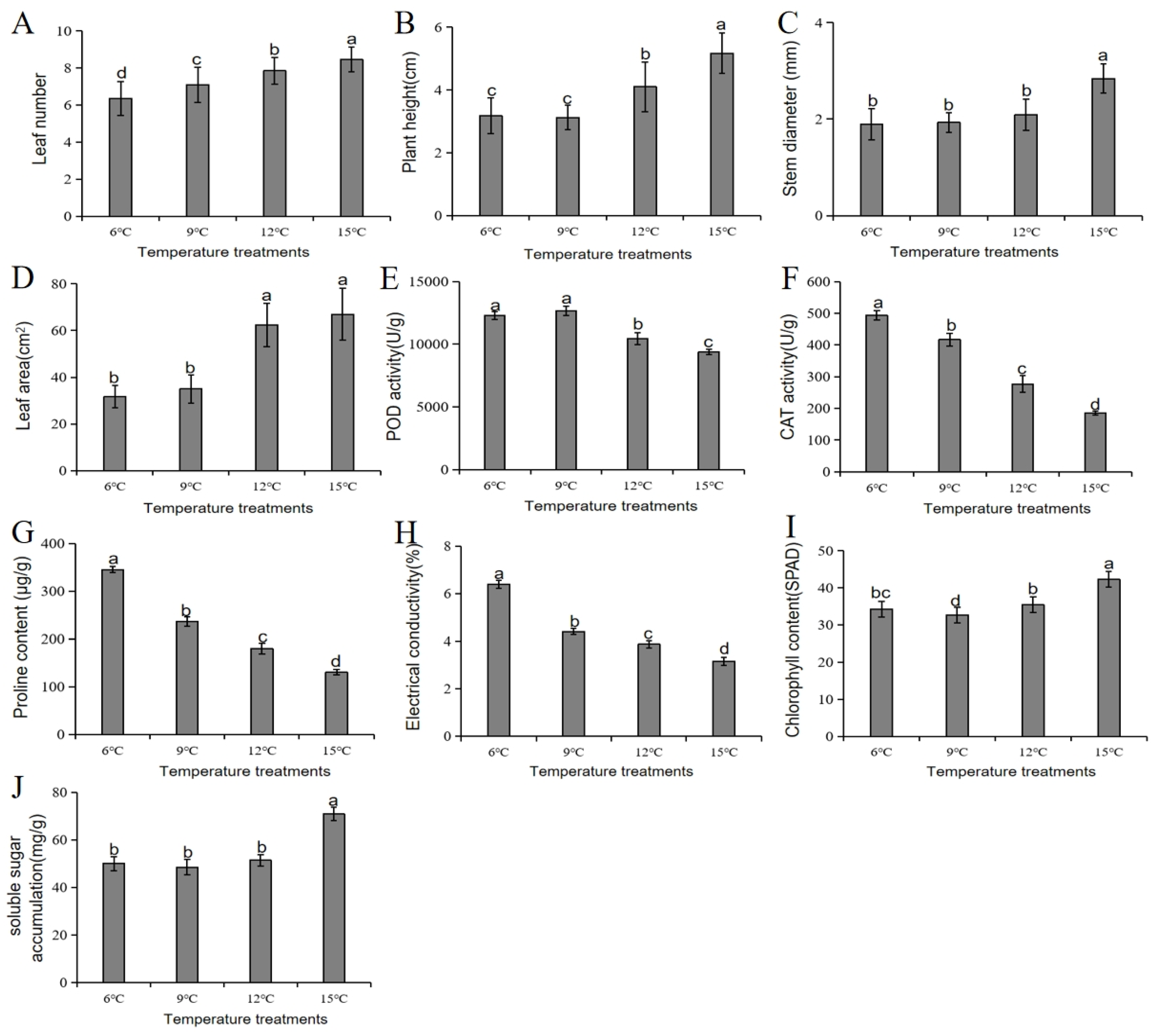
2.2. Phenotypic and Physiological Characterizations
2.3. Total RNA Extraction, cDNA Library Construction, and Transcriptome Analysis
2.4. Sequencing Reads Processing, Genome Mapping, and Gene Expression Quantification
2.5. Differentially Expressed Genes (DEGs) Detection, Functional Enrichment Analysis, and Identification of Key Cold-Responsive DEGs
2.6. Quantitative Real Time-PCR (qRT-PCR) Analysis
2.7. Statistical Analysis of Physiological Data
3. Results
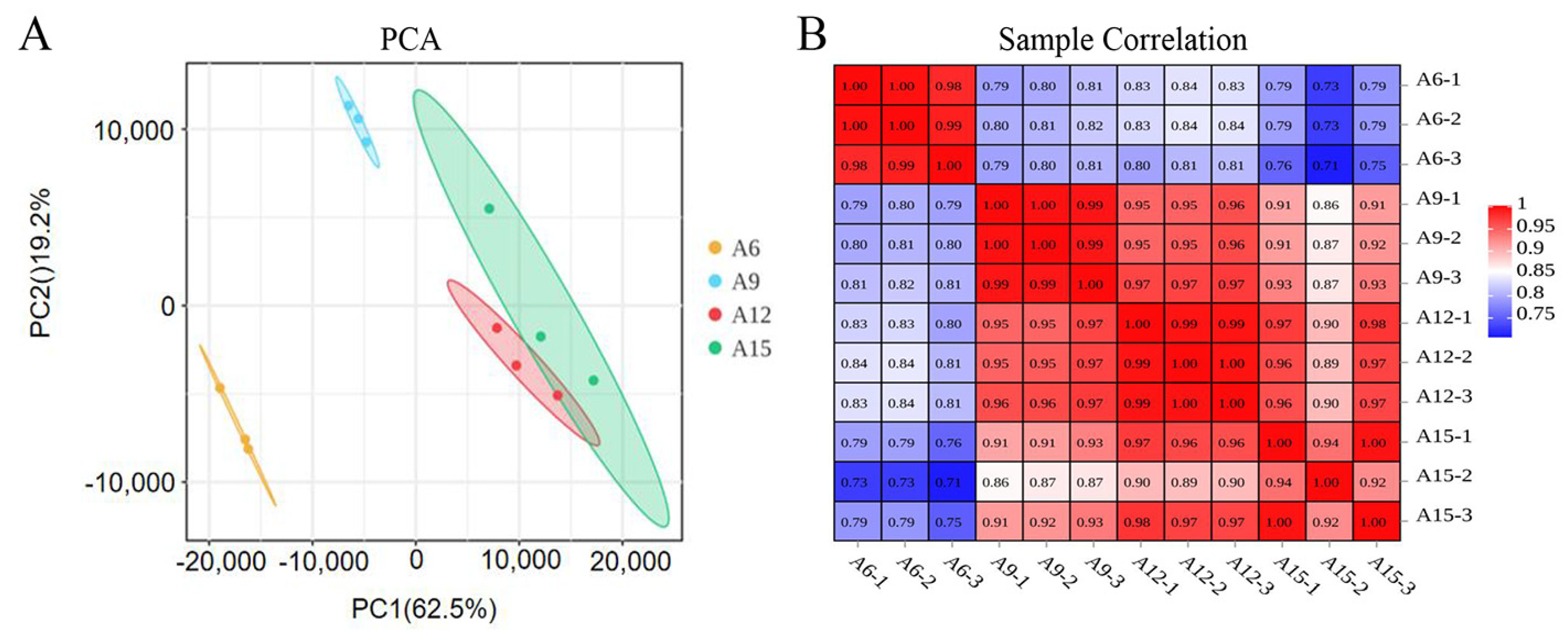
3.1. Summary and Quality Assessment of RNA-Seq Results
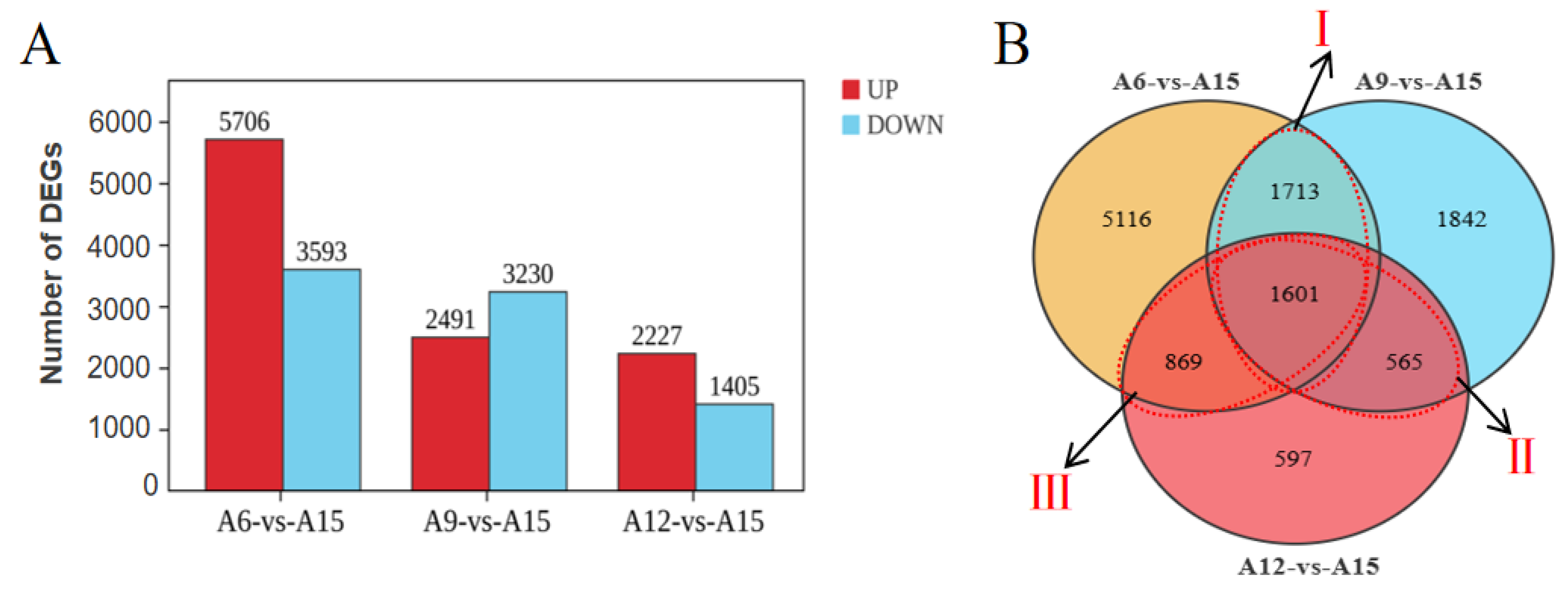
3.2. Gene Differential Expression Analysis
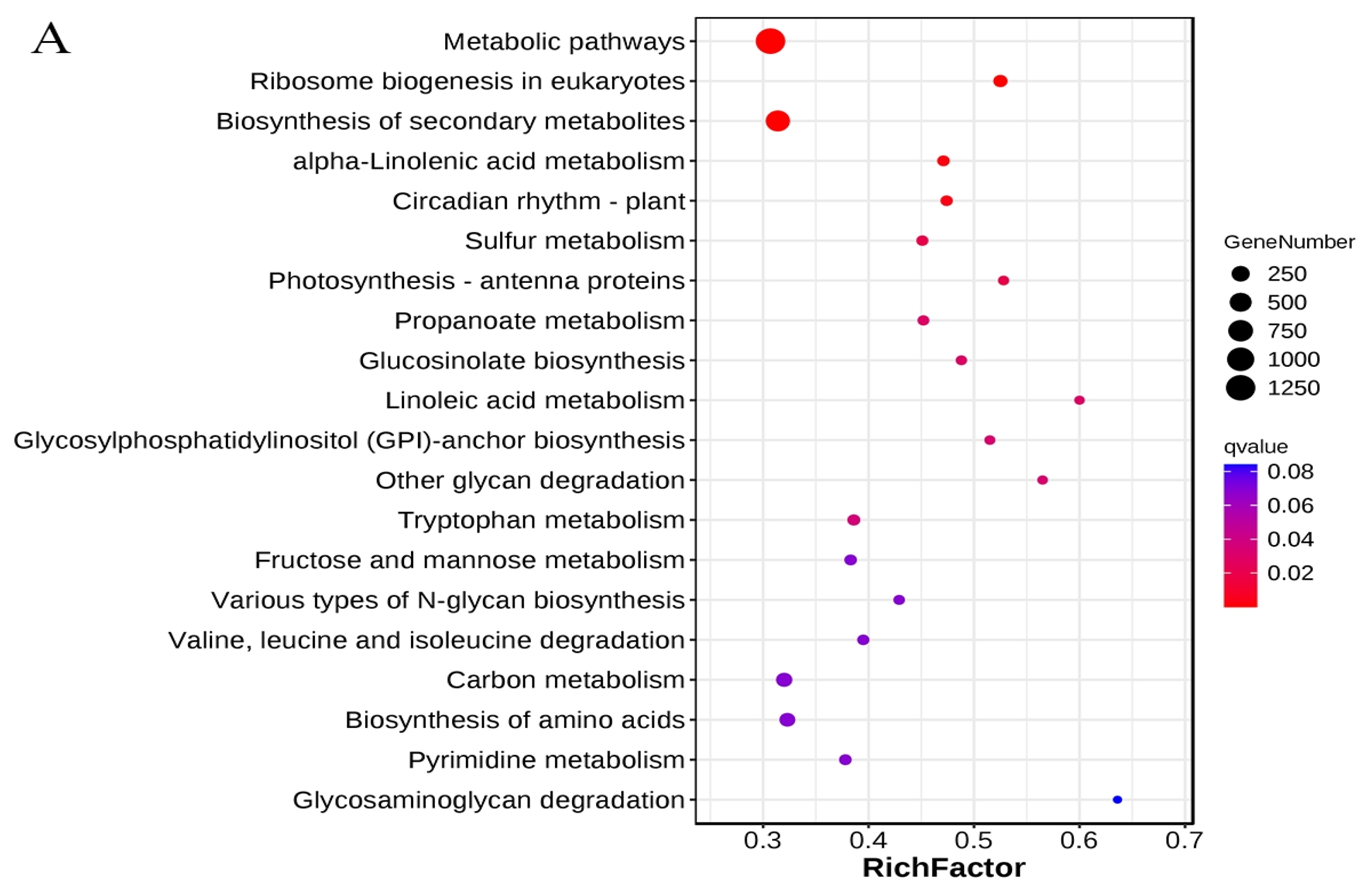
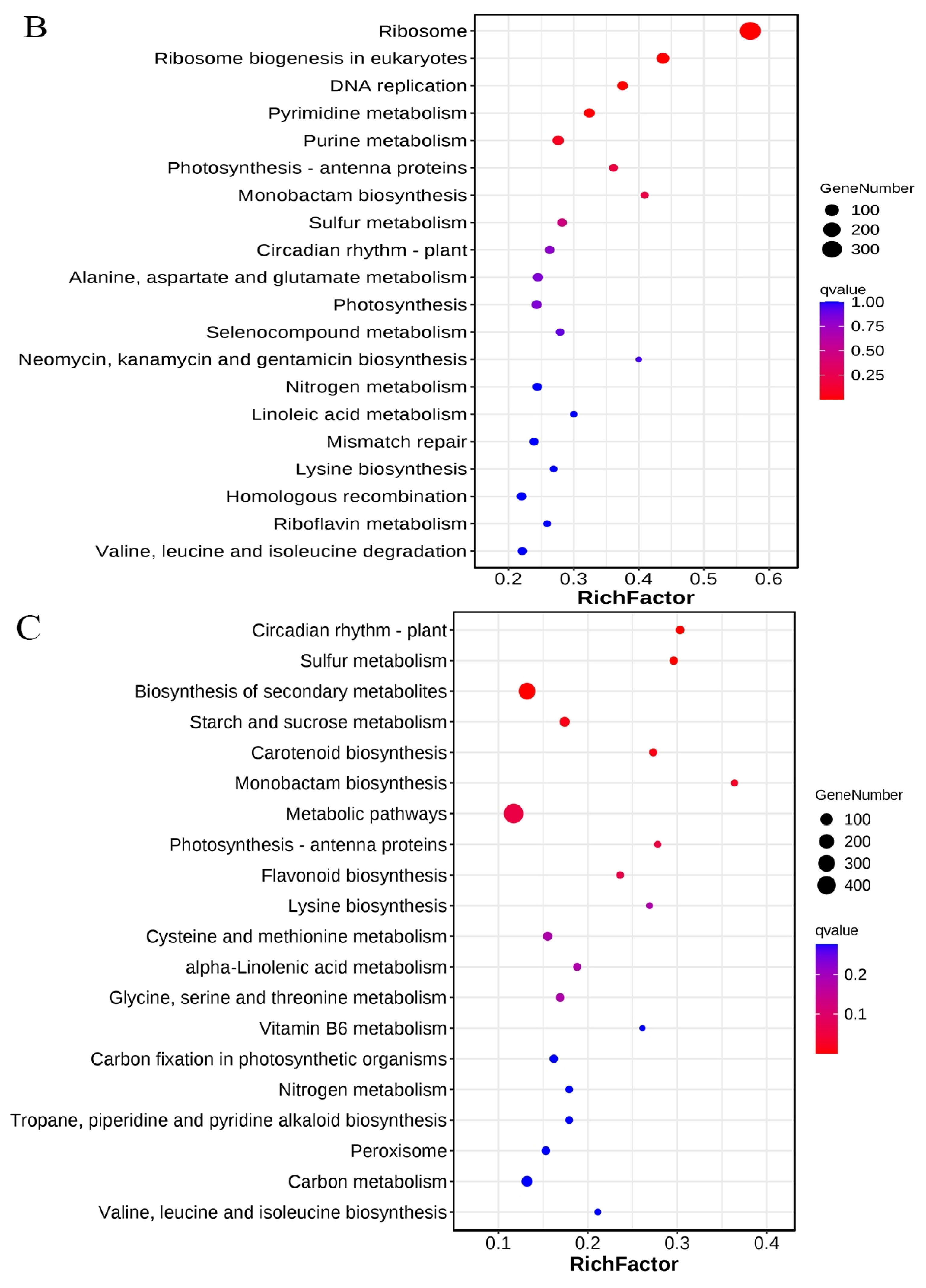
3.3. KEGG Metabolic Pathways Enrichment Analysis of the DEGs
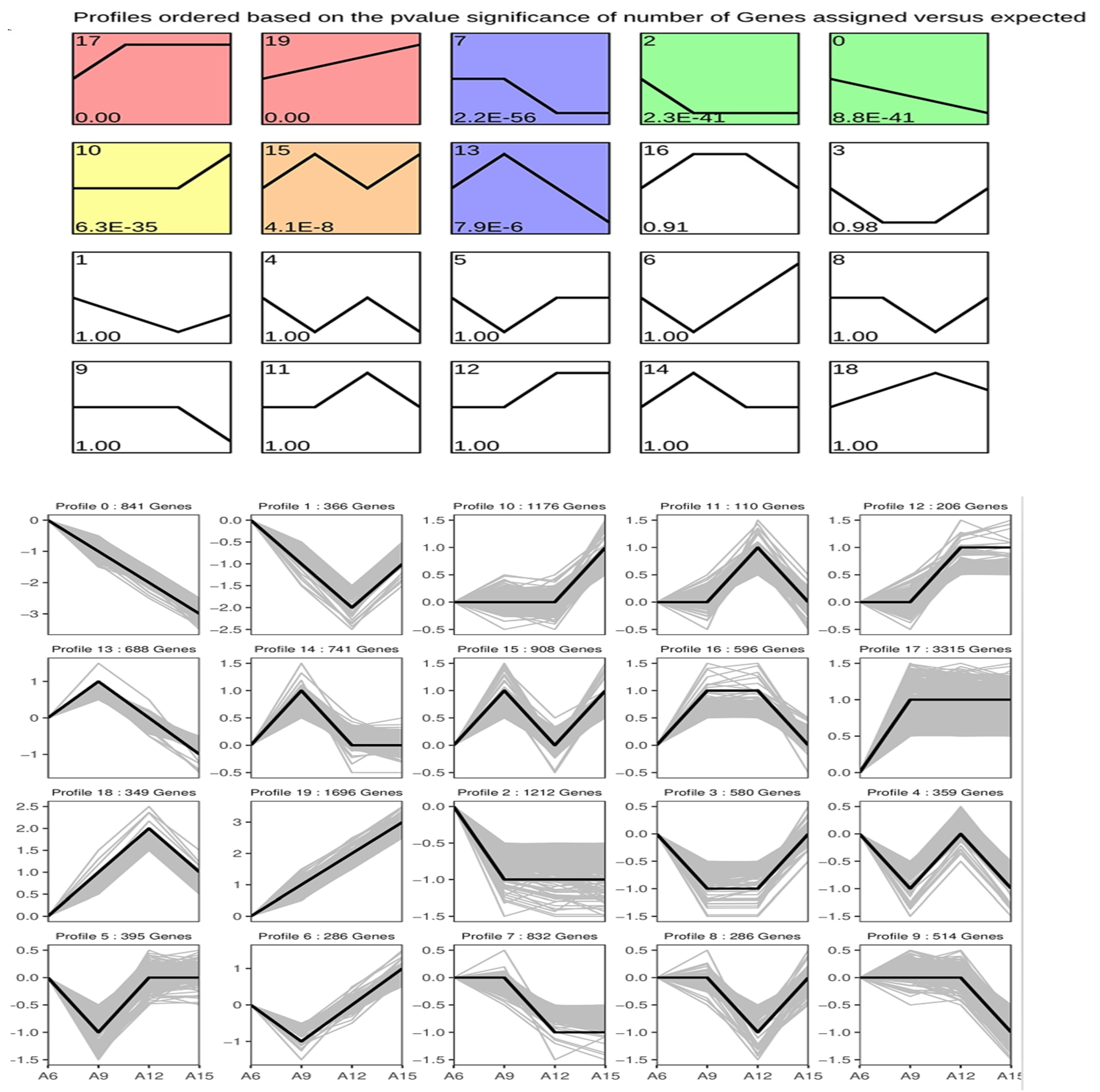
3.4. K-Means Clustering Analysis of DEGs
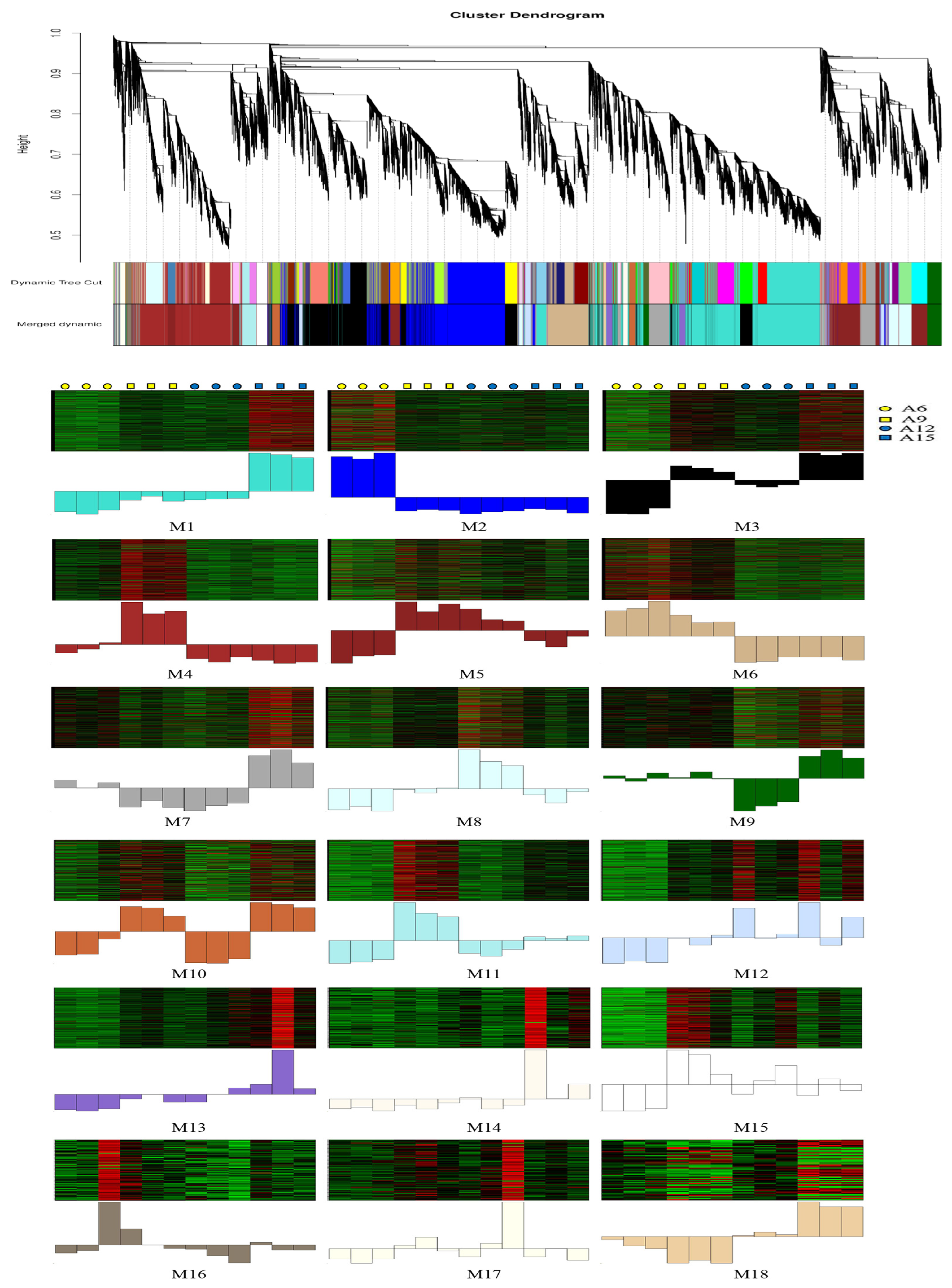
3.5. WGCNA of Differentially Expressed Genes
3.6. Identification of Hub Genes Associated with Vernalization
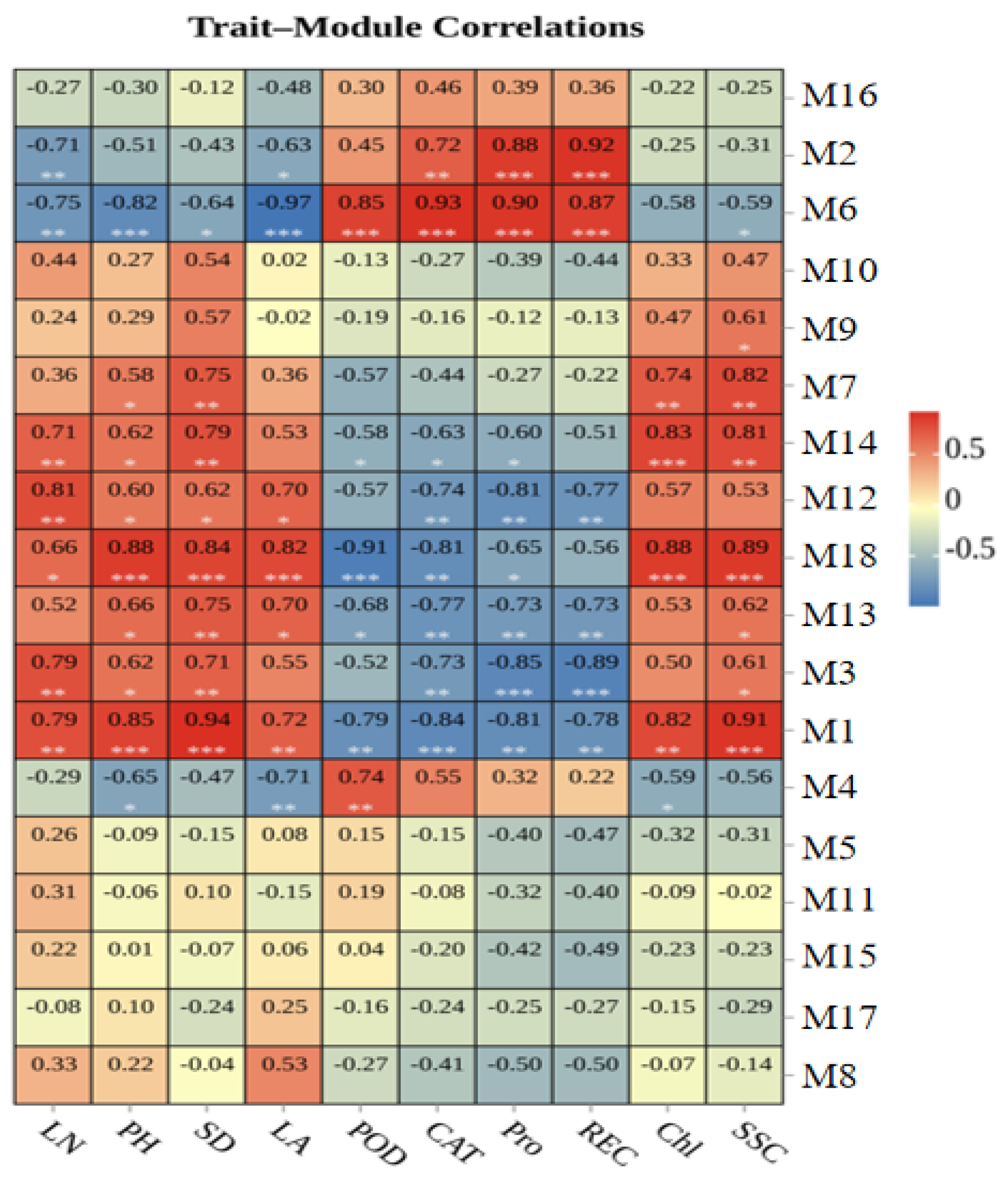
3.7. Association of Modules with Phenotypic and Physiological Traits
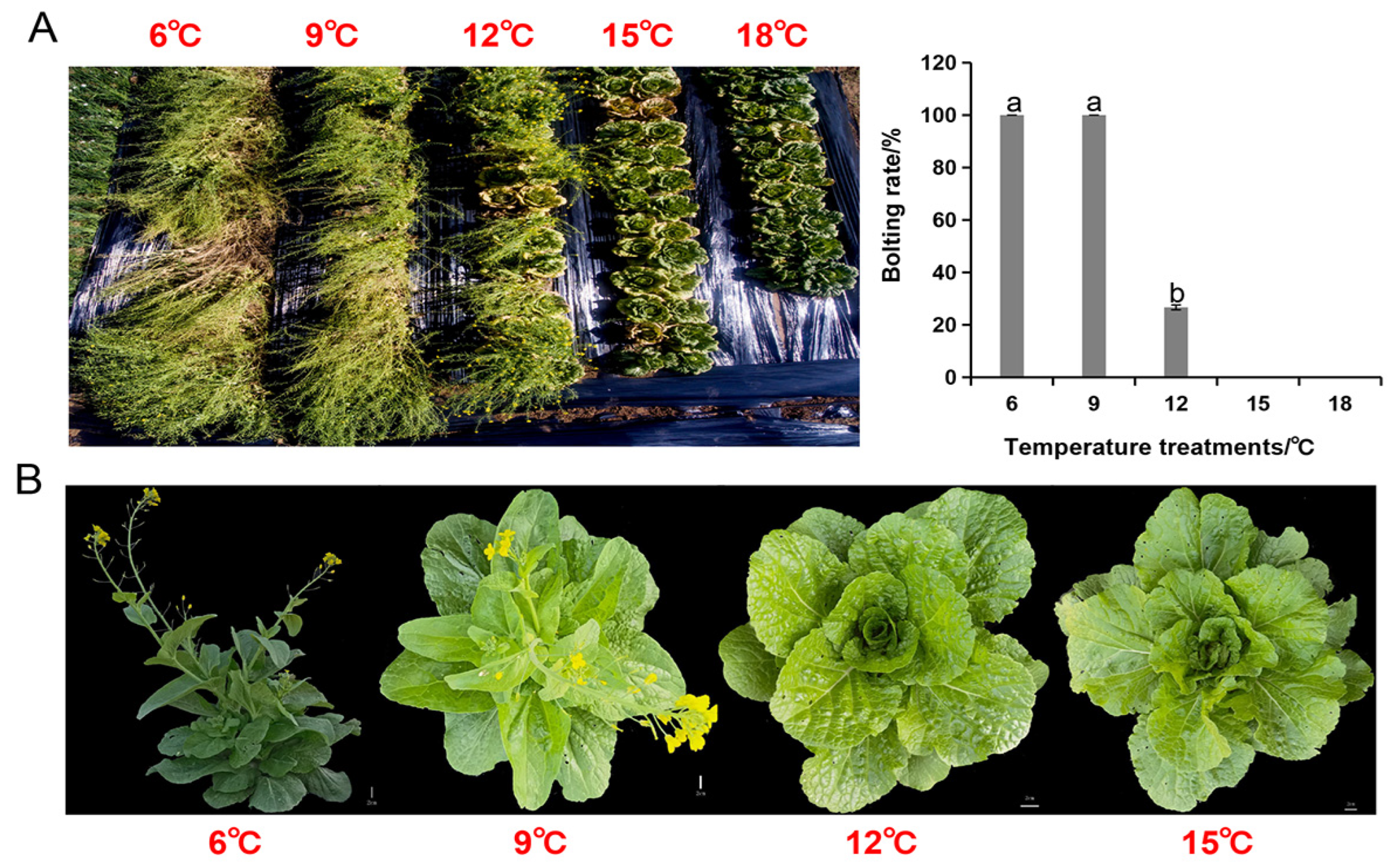
3.8. Phenotypic and Physiological Responses of Chinese Cabbage to Cold Stress at Different Temperature Treatments
3.9. Quantitative Real-Time PCR (qRT-PCR) Validation
4. Discussion
4.1. Chinese Cabbage Differential Responses to Low Temperature (Cold Stress) Gradient at the Phenotypic and Physiological Levels
4.2. Key Cold-Stress-Responsive (Hub) Genes Identified by Transcriptome Analysis and WGCNA
4.3. Analysis of Module–Trait Association and the Expression Patterns of Key Cold-Responsive Genes Identified in the Special Modules
4.4. DEGs Related to ‘Response to Abiotic Stress’ Identified Under Cold Conditions
4.5. Significantly Enriched Metabolic Pathways of DEGs Under Cold Stress
4.6. Nonlinear Transcriptomic Responses to Moderate Cold Stress Reveal Complex Regulatory Mechanisms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tian, J.; Zhang, C.; Chen, X.; Liu, Y.; Zhao, J.; Wu, Q. Brassica rapa L. ssp. pekinensis (Chinese cabbage): A review of the plant’s cultivation, genetic diversity, and health benefits. J. Agric. Sci. 2012, 150, 321–329. [Google Scholar]
- Zhang, L.; Wang, Y.; Liu, J.; Zhou, X.; Chen, S.; Sun, F. The effect of temperature on the growth and bolting behavior of Chinese cabbage. Hortic. Sci. 2017, 52, 35–43. [Google Scholar]
- Wang, X.; Chen, Z.; Zhang, D.; Liu, J.; Li, F.; Sun, Y. Advances in preventing bolting in Chinese cabbage: Strategies and techniques. Acta Hortic. 2015, 1090, 231–238. [Google Scholar]
- Liu, R.; Xie, J.; Li, Y.; Chen, M.; Xu, T.; Huang, F. Health benefits of Brassica rapa ssp. pekinensis: Antioxidant properties and phytochemicals. Food Chem. 2018, 240, 1056–1065. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, G.; Xu, T.; Chen, Y.; Li, S.; Han, R. Cold-induced sugar accumulation and quality improvement in Chinese cabbage. Hortic. Plant J. 2019, 5, 213–220. [Google Scholar]
- Wang, X.; Zhang, L.; Chen, Y.; Huang, B.; Zhao, S.; Wang, Z. Bolting in Chinese cabbage under cold stress: Mechanisms and control strategies. Plant Sci. 2017, 256, 46–54. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Li, W.; Zhang, X.; Yu, D.; Xu, L. Breeding cold-tolerant Chinese cabbage for winter cultivation: Recent advances. Agric. Sci. J. 2021, 72, 359–367. [Google Scholar]
- Li, J.; Sun, Y.; Yang, W.; Huang, X.; Wu, J.; Zhang, Q. Cold stress responses in Chinese cabbage: Physiological and genetic insights. Plant Physiol. Biochem. 2020, 157, 14–22. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, J.; Chen, H.; Yu, B.; Zhao, Z.; Li, X. Effects of vernalization on bolting and flowering in Chinese cabbage. Hortic. Sci. 2015, 50, 157–163. [Google Scholar]
- Sun, X.; Zhang, L.; Li, X.; Chen, Y.; Zhao, Z.; Liu, H. Advances in seedling transplantation techniques to control bolting in Chinese cabbage. Sci. Hortic. 2014, 165, 107–113. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, J.; Xie, Z.; Tang, C.; Wu, Y.; Zheng, Q. Physiological and biochemical responses of Chinese cabbage to low-temperature vernalization. Plant Biol. Rep. 2018, 36, 455–462. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B.; Snyder, M. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wu, X.; Liu, H.; Li, J.; Zhang, Z.; Zhao, F. RNA-Seq for gene expression profiling and biomarker discovery in agriculture and environmental studies. Plant Biotechnol. J. 2015, 13, 156–167. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, J.; Li, S. Differential transcriptome profiling of chilling stress response between shoots and rhizomes of Oryza longistaminata using RNA sequencing. PLoS ONE 2017, 12, e0188625. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Zhang, Z.; Xie, Y. Comparative transcriptome analysis reveals molecular response to salinity stress of salt-tolerant and sensitive genotypes of indica rice at seedling stage. Sci. Rep. 2018, 8, 2085. [Google Scholar] [CrossRef] [PubMed]
- Zenda, T.; Liu, S.; Wang, X.; Liu, G.; Jin, H.; Dong, A.; Yang, Y.; Duan, H. Key maize drought-responsive genes and pathways revealed by comparative transcriptome and physiological analyses of contrasting inbred lines. Int. J. Mol. Sci. 2019, 20, 1268. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zhang, Z.; Zhao, X. Hub gene mining and co-expression network construction of low-temperature response in maize of seedling by WGCNA. Genes 2023, 14, 1598. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Sun, X.; Wang, C.; Li, F.; Zhang, S.; Zhang, H.; Li, G.; Yuan, L.; Chen, G.; Sun, R.; et al. Gene co-expression network analysis reveals key pathways and hub genes in Chinese cabbage (Brassica rapa L.) during vernalization. BMC Genom. 2021, 22, 236. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhou, J.; Zhang, B.; Zheng, D.; Wang, K.; Han, J. Time-course transcriptome analysis reveals gene co-expression networks and transposable element responses to cold stress in cotton. BMC Genom. 2025, 26, 235. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Pan, Y.-B.; Su, Y.; Zou, W.; Xu, F.; Sun, T.; Grisham, M.P.; Yang, S.; Xu, L.; Que, Y. WGCNA identifies a comprehensive and dynamic gene co-expression network that associates with smut resistance in sugarcane. Int. J. Mol. Sci. 2022, 23, 10770. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, S.; Sun, X.; Li, G.; Yuan, L.; Li, F.; Zhang, H.; Zhang, S.; Chen, G.; Wang, C.; et al. Comparative transcriptome analysis of gene expression and regulatory characteristics associated with different vernalization periods in Brassica rapa. Genes 2020, 11, 392. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Nugroho, A.B.D.; Park, M.; Kim, J.A.; Lee, S.W.; Moon, H.; Choi, D.; Kim, S.; Kim, D.-H. Vernalization regulates flowering genes and modulates glucosinolates biosynthesis in Chinese cabbage. J. Plant Biol. 2022, 65, 157–173. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Rouse, D.T.; Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. The molecular basis of vernalization: The central role of FLOWERING LOCUS C (FLC). Proc. Natl. Acad. Sci. USA 2000, 97, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Xu, M.; Zhang, X.; Liu, Y. Vernalization can regulate flowering time through microRNA mechanism in Brassica rapa. Physiol. Plant. 2018, 164, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ding, Y.; Yang, S. Molecular Regulation of CBF Signaling in Cold Acclimation. Trends Plant Sci. 2018, 23, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhang, H.; Zhang, B.; Wu, X.; Shao, S.; Li, Y.; Hou, X.; Liu, T. Role of vernalization-mediated demethylation in the floral transition of Brassica rapa. Planta 2017, 245, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Sun, C.; He, Y. Epigenetics of plant vernalization regulated by non-coding RNAs. Hereditas 2012, 34, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Yemm, E.W.; Willis, A.J. The estimation of carbohydrates in plant extracts by anthrone. Biochem. J. 1954, 57, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Liu, G.; Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Dong, A.; Yang, Y.; Duan, H. Comparative transcriptomic and physiological analyses of contrasting hybrid cultivars ND476 and ZX978 identify important differentially expressed genes and pathways regulating drought stress tolerance in maize. Genes Genom. 2020, 42, 937–955. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, X.; Wei, S.; Wang, H.; Xu, Y.; Zhao, P. Thermal regulation of carbohydrate metabolism and cell expansion in Brassica rapa: Implications for cold tolerance and yield improvement. Sci. Hortic. 2022, 304, 111298. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Liu, Y.; Zhang, J.; Hu, S.; Zhou, Y. Chilling stress responses in leafy vegetables: Physiological inhibition of meristem activity and nutrient partitioning. Front. Plant Sci. 2021, 12, 670893. [Google Scholar]
- Hussain, S.; Khan, F.; Li, J.; Rauf, A.; Hussain, I.; Maqbool, M. Cold stress-induced oxidative damage and antioxidant defense mechanisms in Brassica crops: A focus on membrane stability and ROS scavenging. Plant Physiol. Rep. 2020, 25, 321–334. [Google Scholar] [CrossRef]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R.; Hasegawa, P. Resource allocation trade-offs in plants under abiotic stress: From photosynthetic priming to growth-defense balance. Trends Plant Sci. 2022, 27, 786–801. [Google Scholar] [CrossRef]
- Shi, F.; Xu, H.; Liu, C.; Tan, C.; Ren, J.; Ye, X.; Feng, H.; Liu, Z. Whole-transcriptome sequencing reveals a vernalization-related ceRNA regulatory network in Chinese cabbage (Brassica campestris L. ssp. pekinensis). BMC Genom. 2021, 22, 819. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeong, J.; Choi, S.; Park, Y.; Seo, H.; Park, C. Regulation of the expression of Rubisco activase in response to low temperature and light in plants. Plant Physiol. Biochem. 2015, 96, 369–376. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, K.P.; Liu, Y.; Kim, C. Temperature-driven changes in membrane fluidity differentially impact FILAMENTATION TEMPERATURE-SENSITIVE H2-mediated photosystem II repair. Plant Cell 2024, 37, koae323. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Allakhverdiev, S.I.; Yamamoto, H.; Hayashi, H.; Murata, N.; Yokota, A. Photoinhibition-repair cycle protects photosystem II from long-term photodamage under environmental stress. Plant Cell Physiol. 2006, 47, 506–514. [Google Scholar] [CrossRef]
- Kong, X.; Wei, B.; Gao, Z.; Zhou, Y.; Shi, F.; Zhou, X.; Zhou, Q.; Ji, S. Changes in Membrane Lipid Composition and Function Accompanying Chilling Injury in Bell Peppers. Plant Cell Physiol. 2018, 59, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Shi, Y.; Yang, S.; Gong, Z.; Zhu, J.; Guo, H. Cold stress in plants: A comprehensive review on the molecular regulators involved in cold acclimation. J. Plant Biol. 2019, 62, 301–310. [Google Scholar]
- Guo, X.; Liu, D.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, Y.; Yang, Y.; Song, C.; Wang, B.; Yang, S.; Guo, Y.; Gong, Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jia, Y.; Ding, Y.; Shi, Y.; Li, Z.; Guo, Y.; Gong, Z.; Yang, S. Plasma Membrane CRPK1-Mediated Phosphorylation of 14-3-3 Proteins Induces Their Nuclear Import to Fine-Tune CBF Signaling during Cold Response. Mol. Cell 2017, 66, 117–128.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dang, P.; Liu, L.; He, C. Cold acclimation by the CBF-COR pathway in a changing climate: Lessons from Arabidopsis thaliana. Plant Cell Rep. 2019, 38, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, M.; Satoh, R.; Maruyama, K.; Parvez, M.M.; Seki, M.; Hiratsu, K.; Ohme-Takagi, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 2013, 17, 3470–3488. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Z.; Zhang, Y.; Guo, J.; Liu, L.; Wang, C.; Wang, B.; Han, G. The roles of HD-ZIP proteins in plant abiotic stress tolerance. Front. Plant Sci. 2022, 13, 1027071. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, N.; Chew, W.; Sornaraj, P.; Borisjuk, N.; Yang, N.; Singh, R.; Bazanova, N.; Shavrukov, Y.; Guendel, A.; Munz, E.; et al. The homeodomain transcription factor TaHDZipI-2 from wheat regulates frost tolerance, flowering time and spike development in transgenic barley. New Phytol. 2016, 211, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Ding, Y.; Hu, S.; Ding, L.; Chen, Z.; Zhu, C. The role of HD-zip class I transcription factors in plant response to abiotic stresses. Physiol. Plant. 2019, 167, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Dubos, C. The Arabidopsis bHLH transcription factor family. Trends Plant Sci. 2024, 29, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Kim, H.; Yu, T.; Zhang, A.; Yang, Y.; Liu, M.; Yu, W.; Zhao, P.; Zhang, Q.; Cao, Q.; et al. Identification and function analysis of bHLH genes in response to cold stress in sweetpotato. Plant Physiol. Biochem. 2021, 169, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Fernando, V.C.D. Cross Talk Between Cold Stress Response Signaling Pathway and Other Stress Response Pathways. In Cold Tolerance in Plants; Wani, S., Herath, V., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Knight, H.; Zarka, D.; Okamoto, H.; Thomashow, M.F.; Knight, M.R. Abscisic acid induces CBF gene transcription and subsequent induction of cold-regulated genes via the CRT promoter element. Plant Physiol. 2004, 135, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Javed, T.; Liu, T.; Ali, A.; Gao, J. Mechanisms of Abscisic acid (ABA)-mediated plant defense responses: An updated review. Plant Stress 2025, 15, 100724. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed]
- Zipor, G.; Oren-Shamir, M. Do vacuolar peroxidases act as plant caretakers? Plant Sci. 2013, 199, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.K.; Sunkar, R. Gene regulation during cold stress acclimation in plants. Methods Mol. Biol. 2010, 639, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Chakrabarty, D. Dehydrin in the past four decades: From chaperones to transcription co-regulators in regulating abiotic stress response. Curr. Res. Biotechnol. 2021, 3, 249–259. [Google Scholar] [CrossRef]
- Fujita, Y.; Fujita, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 2011, 124, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.F. Molecular basis of plant cold acclimation: Insights gained from studying the CBF cold response pathway. Plant Physiol. 2010, 154, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Liberek, K.; Lewandowska, A.; Ziętkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 2008, 27, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Hao, Z.; Sun, Z.; Zheng, X.; Wang, M.; Zhang, Y. Heat shock proteins and abiotic stress tolerance in plants. Front. Biol. 2012, 7, 326–335. [Google Scholar]
- Winkel-Shirley, B. Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Biol. 2002, 5, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Salam, U.; Ullah, S.; Tang, Z.H.; Elateeq, A.A.; Khan, Y.; Khan, J.; Khan, A.; Ali, S. Plant Metabolomics: An Overview of the Role of Primary and Secondary Metabolites against Different Environmental Stress Factors. Life 2023, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Kanwal, F.; Ullah, S.; Fahad, M.; Tariq, L.; Altaf, M.T.; Riaz, A.; Zhang, G. Plant Secondary Metabolites—Central Regulators Against Abiotic and Biotic Stresses. Metabolites 2025, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Das, G.; Das, S.K.; Chakraborty, A.; Banerjee, R.; Jana, S. Roles of flavonoids in plants. Int. J. Pharm. Sci. Technol. 2011, 6, 12–35. [Google Scholar]
- Li, J.; Yu, Q.; Liu, C.; Zhang, N.; Xu, W. Flavonoids as key players in cold tolerance: Molecular insights and applications in horticultural crops. Hortic. Res. 2025, 12, uhae366. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.; Kopka, J.; Sung, D.Y.; Zhao, W.; Popp, M.; Porat, R. Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol. 2007, 136, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Beckles, D.M. Dynamic changes in the starch-sugar interconversion within plant source and sink tissues promote a better abiotic stress response. J. Plant Physiol. 2019, 234–235, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, M.; Santelia, D.; Fenske, R.; Trevisan, M.; Viola, R.; Zeeman, S.C. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017, 214, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, J.; Duan, X.; Ding, W.; Xu, M.; Xiong, Y. Target of Rapamycin (TOR): A Master Regulator in Plant Growth, Development, and Stress Responses. Annu. Rev. Plant Biol. 2025, 76, 341–371. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads (%) | Unique_Mapped (%) | Multiple_Mapped (%) | AF_Q30 (%) | AF_GC (%) |
|---|---|---|---|---|---|---|
| A6-1 | 42,083,140 | 41,870,984 (99.50%) | 37,672,913 (90.24%) | 998,937 (2.39%) | 5,791,987,638 (92.70%) | 2,988,649,887 (47.84%) |
| A6-2 | 43,646,058 | 43,468,442 (99.59%) | 39,461,219 (90.99%) | 1,074,605 (2.48%) | 6,073,563,050 (93.67%) | 3,098,203,406 (47.78%) |
| A6-3 | 41,728,384 | 41,555,956 (99.59%) | 37,723,296 (90.95%) | 1,029,352 (2.48%) | 5,840,601,159 (94.10%) | 2,947,541,057 (47.49%) |
| A9-1 | 43,316,474 | 43,129,816 (99.57%) | 38,905,260 (90.40%) | 1,019,417 (2.37%) | 6,028,558,747 (93.64%) | 3,056,572,130 (47.48%) |
| A9-2 | 46,400,400 | 46,207,588 (99.58%) | 41,657,360 (90.39%) | 1,097,035 (2.38%) | 6,479,754,563 (93.93%) | 3,279,608,319 (47.54%) |
| A9-3 | 49,023,646 | 48,804,126 (99.55%) | 44,137,405 (90.66%) | 1,171,789 (2.41%) | 6,781,477,998 (93.13%) | 3,464,902,473 (47.58%) |
| A12-1 | 45,255,406 | 45,082,436 (99.62%) | 40,820,878 (90.84%) | 1,238,429 (2.76%) | 6,270,007,583 (93.18%) | 3,232,348,084 (48.04%) |
| A12-2 | 37,078,896 | 36,942,084 (99.63%) | 33,560,542 (91.11%) | 992,795 (2.70%) | 5,171,610,430 (93.78%) | 2,640,163,034 (47.88%) |
| A12-3 | 47,484,122 | 47,269,836 (99.55%) | 42,801,187 (90.84%) | 1,282,488 (2.72%) | 6,595,279,562 (93.51%) | 3,379,144,523 (47.91%) |
| A15-1 | 43,695,692 | 43,510,072 (99.58%) | 39,363,393 (90.68%) | 1,200,329 (2.77%) | 6,097,045,076 (93.84%) | 3,102,716,312 (47.75%) |
| A15-3 | 53,561,126 | 53,324,268 (99.56%) | 48,169,587 (90.58%) | 1,619,417 (3.05%) | 7,451,366,401 (93.60%) | 3,805,927,059 (47.81%) |
| Module | TF Number | Module | TF Number |
|---|---|---|---|
| turquoise (M1) | 227 | sienna3 (M10) | 28 |
| blue (M2) | 209 | paleturquoise (M11) | 27 |
| black (M3) | 189 | lightsteelblue1 (M12) | 24 |
| brown (M4) | 128 | mediumpurple3 (M13) | 19 |
| brown4 (M5) | 93 | floralwhite (M14) | 11 |
| tan (M6) | 68 | white (M15) | 8 |
| darkgrey (M7) | 47 | bisque4 (M16) | 8 |
| lightcyan1 (M8) | 43 | ivory (M17) | 6 |
| darkgreen (M9) | 33 | navajowhite2 (M18) | 4 |
| Module | GO ID | GO Enrichment Terms | Gene Numbers | p-Value | FDR |
|---|---|---|---|---|---|
| M1 | GO:0016192 | vesicle-mediated transport | 157 | 1.00 × 10−11 | 1.00 × 10−9 |
| GO:0012505 | endomembrane system | 267 | 1.00 × 10−11 | 1.00 × 10−9 | |
| GO:0005515 | protein binding | 846 | 1.00 × 10−11 | 1.00 × 10−9 | |
| GO:0070647 | protein modification by small protein conjugation or removal | 117 | 1.00 × 10−11 | 1.00 × 10−9 | |
| GO:0005794 | golgi apparatus | 127 | 1.00 × 10−11 | 2.00 × 10−6 | |
| M2 | GO:0044281 | small molecule metabolic process | 504 | 1.00 × 10−11 | 1.00 × 10−8 |
| GO:0009628 | response to abiotic stimulus | 177 | 1.00 × 10−11 | 1.00 × 10−6 | |
| GO:0006811 | ion transport | 233 | 1.00 × 10−11 | 1.60 × 10−5 | |
| GO:0010035 | response to inorganic substance | 102 | 1.00 × 10−11 | 2.40 × 10−5 | |
| M3 | GO:0051179 | localization | 585 | 1.00 × 10−11 | 2.00 × 10−9 |
| GO:0005783 | endoplasmic reticulum | 119 | 1.00 × 10−11 | 2.10 × 10−5 | |
| GO:0006810 | transport | 544 | 1.00 × 10−11 | 2.50 × 10−5 | |
| M4 | GO:0006412 | translation | 454 | 1.00 × 10−11 | 1.00 × 10−9 |
| GO:0043043 | peptide biosynthetic process | 460 | 1.00 × 10−11 | 1.00 × 10−9 | |
| GO:0006518 | peptide metabolic process | 469 | 1.00 × 10−11 | 1.00 × 10−9 | |
| M6 | GO:0042254 | ribosome biogenesis | 107 | 1.00 × 10−11 | 1.00 × 10−9 |
| GO:0022613 | ribonucleoprotein complex biogenesis | 109 | 1.00 × 10−11 | 1.00 × 10−9 | |
| GO:0016072 | rRNA metabolic process | 70 | 1.00 × 10−11 | 1.00 × 10−9 | |
| M7 | GO:0009266 | response to temperature stimulus | 21 | 4.00 × 10−6 | 8.47 × 10−3 |
| GO:0009628 | response to abiotic stimulus | 39 | 1.10 × 10−5 | 1.18 × 10−2 | |
| GO:0019685 | photosynthesis, dark reaction | 4 | 3.20 × 10−5 | 1.76 × 10−2 | |
| GO:0019253 | reductive pentose-phosphate cycle | 4 | 3.20 × 10−5 | 1.76 × 10−2 | |
| GO:0009987 | cellular process | 303 | 1.12 × 10−4 | 4.97 × 10−2 | |
| M8 | GO:0006412 | translation | 74 | 1.00 × 10−11 | 2.00 × 10−5 |
| GO:0006518 | peptide metabolic process | 79 | 1.00 × 10−11 | 9.70 × 10−5 | |
| GO:0006518 | peptide metabolic process | 79 | 1.00 × 10−11 | 9.70 × 10−5 | |
| M10 | GO:0010812 | negative regulation of cell–substrate adhesion | 3 | 2.80 × 10−5 | 1.86 × 10−2 |
| GO:0010810 | regulation of cell–substrate adhesion | 3 | 4.50 × 10−5 | 1.86 × 10−2 | |
| GO:0051270 | regulation of cellular component movement | 5 | 5.20 × 10−5 | 1.86 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Liu, S.; Li, H.; Sun, M.; Yan, B.; Zhang, P.; Zhang, L. Global Transcriptome and Weighted Gene Co-Expression Network Analyses of Cold Stress Responses in Chinese Cabbage. Genes 2025, 16, 845. https://doi.org/10.3390/genes16070845
Zhang J, Liu S, Li H, Sun M, Yan B, Zhang P, Zhang L. Global Transcriptome and Weighted Gene Co-Expression Network Analyses of Cold Stress Responses in Chinese Cabbage. Genes. 2025; 16(7):845. https://doi.org/10.3390/genes16070845
Chicago/Turabian StyleZhang, Jizong, Songtao Liu, Huibin Li, Mengmeng Sun, Baoyue Yan, Peng Zhang, and Lifeng Zhang. 2025. "Global Transcriptome and Weighted Gene Co-Expression Network Analyses of Cold Stress Responses in Chinese Cabbage" Genes 16, no. 7: 845. https://doi.org/10.3390/genes16070845
APA StyleZhang, J., Liu, S., Li, H., Sun, M., Yan, B., Zhang, P., & Zhang, L. (2025). Global Transcriptome and Weighted Gene Co-Expression Network Analyses of Cold Stress Responses in Chinese Cabbage. Genes, 16(7), 845. https://doi.org/10.3390/genes16070845