
Deficiency in KPNA4, but Not in KPNA3, Causes Attention Deficit/Hyperactivity Disorder like Symptoms in Mice

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. Assessment of Motoric Abilities and Locomotion
2.4. Statistical Analysis
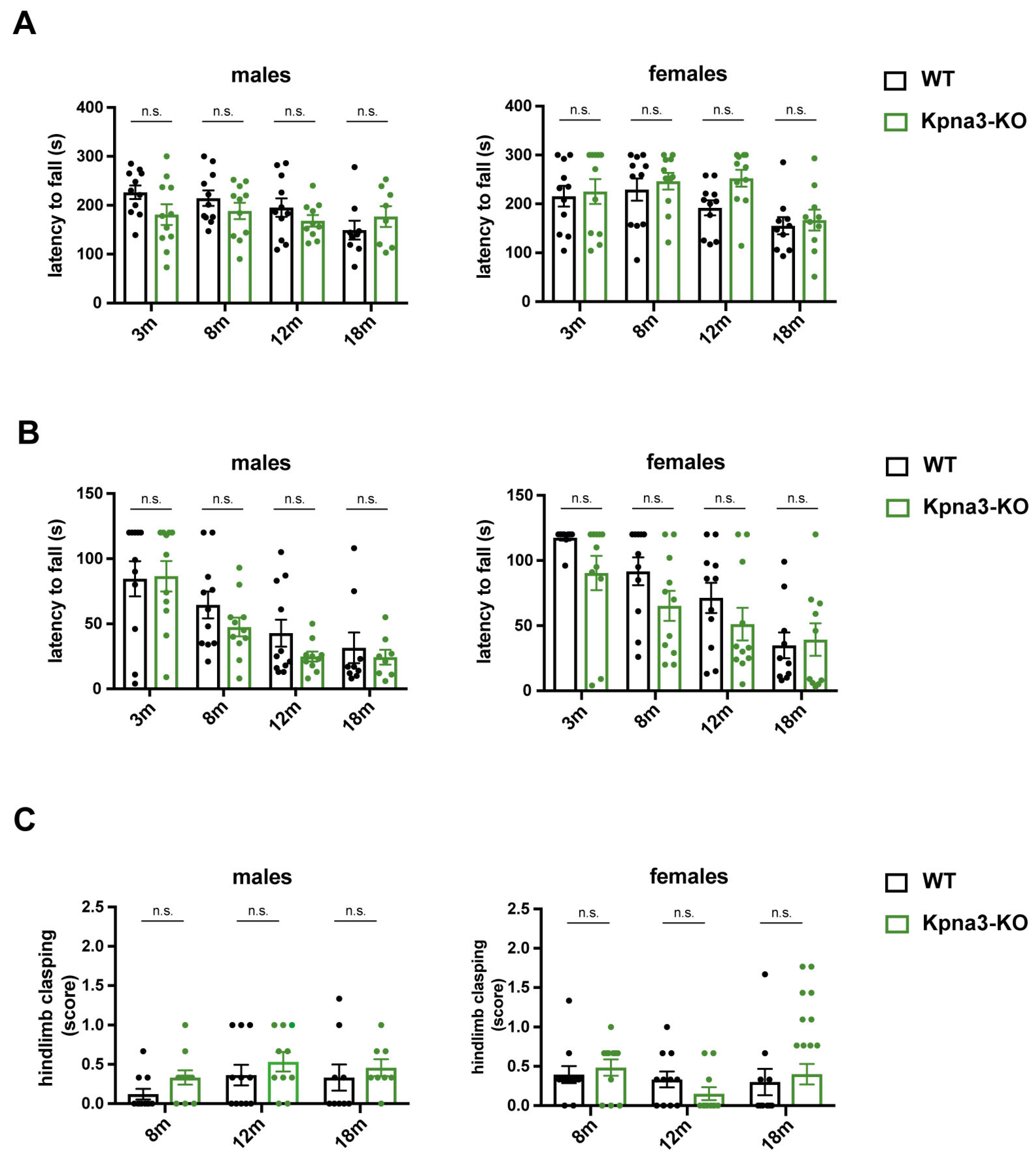
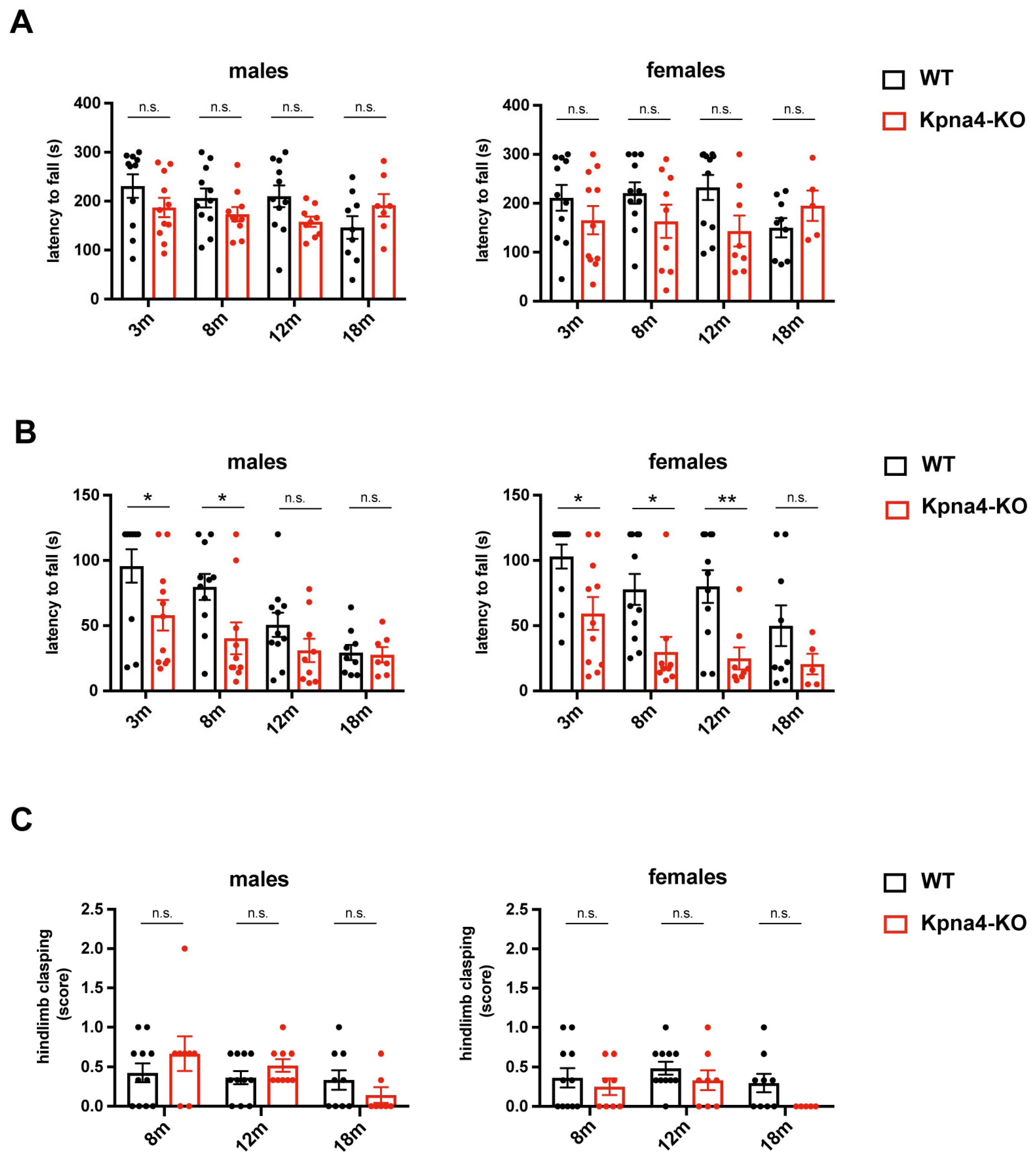
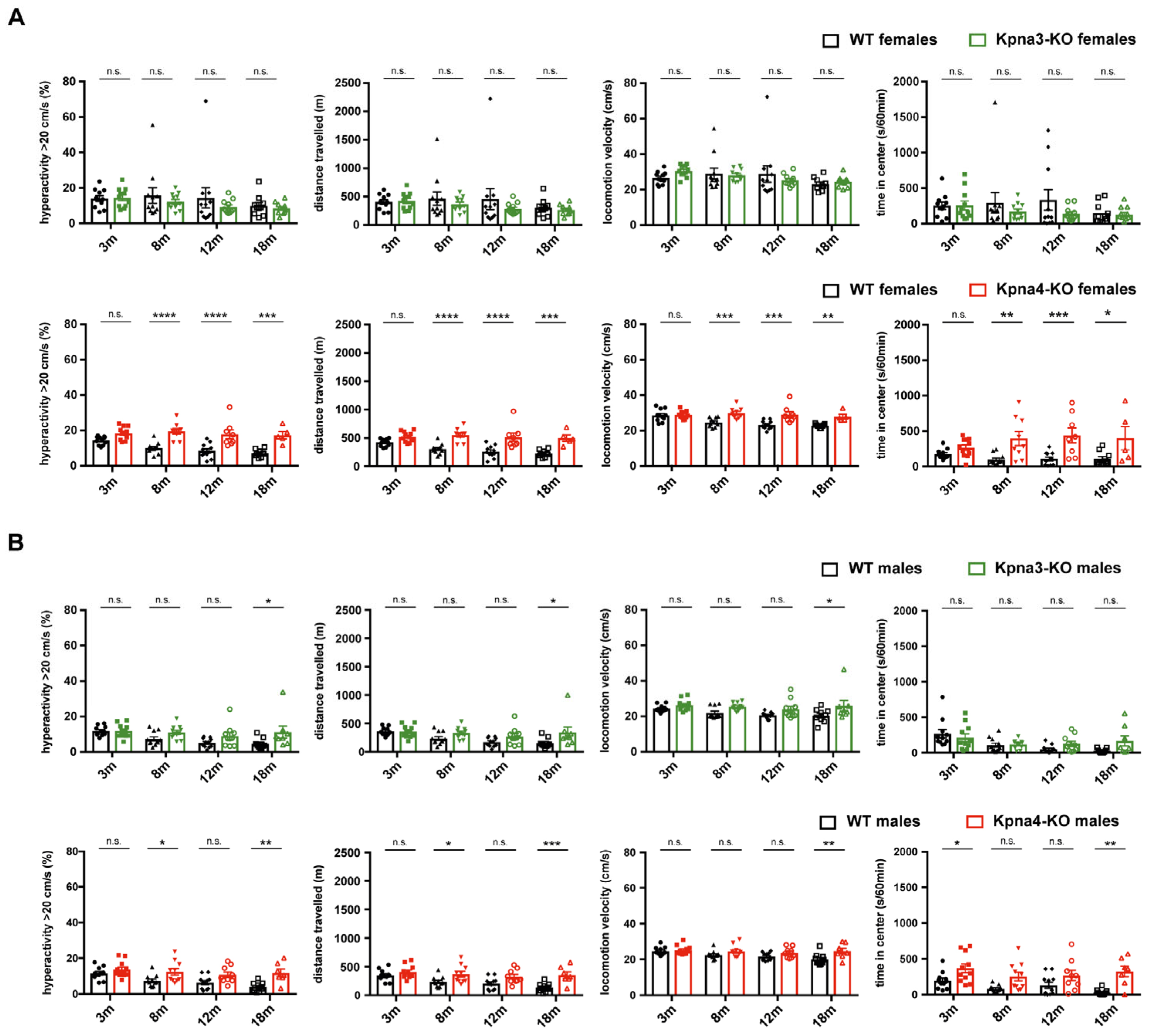
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADHD | Attention deficit/hyperactivity disorder |
| NCT | Nucleocytoplasmic transport |
| ALS | Amyotrophic lateral sclerosis |
| FTD | Frontotemporal dementia (FTD) |
| HSP | Hereditary spastic paraplegia |
| MND | Motor neuron disease |
| WT | Wildtype |
| GWAS-MA | Genome-wide association study metaanalysis |
References
- Plessis-Belair, J.; Russo, T.; Riessland, M.; Sher, R.B. Nuclear Import Defects Drive Cell Cycle Dysregulation in Neurodegeneration. Aging Cell 2025, 16, e70091. [Google Scholar] [CrossRef]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Prpar Mihevc, S.; Darovic, S.; Kovanda, A.; Bajc Cesnik, A.; Zupunski, V.; Rogelj, B. Nuclear trafficking in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain 2017, 140, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Rothstein, J.D. Nuclear pore complexes—A doorway to neural injury in neurodegeneration. Nat. Rev. Neurol. 2022, 18, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Ito, H.; Hirano, A.; Fujita, K.; Wate, R.; Nakamura, M.; Kaneko, S.; Nakano, S.; Kusaka, H. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 1184–1192. [Google Scholar] [CrossRef]
- Solomon, D.A.; Stepto, A.; Au, W.H.; Adachi, Y.; Diaper, D.C.; Hall, R.; Rekhi, A.; Boudi, A.; Tziortzouda, P.; Lee, Y.B.; et al. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-α mediates C9orf72-related neurodegeneration. Brain 2018, 141, 2908–2924. [Google Scholar] [CrossRef]
- Xiao, S.; MacNair, L.; McGoldrick, P.; McKeever, P.M.; McLean, J.R.; Zhang, M.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 78, 568–583. [Google Scholar] [CrossRef]
- Kohler, M.; Ansieau, S.; Prehn, S.; Leutz, A.; Haller, H.; Hartmann, E. Cloning of two novel human importin-α subunits and analysis of the expression pattern of the importin-α protein family. FEBS Lett. 1997, 417, 104–108. [Google Scholar] [CrossRef]
- Tsuji, L.; Takumi, T.; Imamoto, N.; Yoneda, Y. Identification of novel homologues of mouse importin α, the α subunit of the nuclear pore-targeting complex, and their tissue-specific expression. FEBS Lett. 1997, 416, 30–34. [Google Scholar] [CrossRef]
- Tejomurtula, J.; Lee, K.B.; Tripurani, S.K.; Smith, G.W.; Yao, J. Role of importin alpha8, a new member of the importin α family of nuclear transport proteins, in early embryonic development in cattle. Biol. Reprod. 2009, 81, 333–342. [Google Scholar] [CrossRef]
- Kohler, M.; Speck, C.; Christiansen, M.; Bischoff, F.R.; Prehn, S.; Haller, H.; Gorlich, D.; Hartmann, E. Evidence for distinct substrate specificities of importin α family members in nuclear protein import. Mol. Cell Biol. 1999, 19, 7782–7791. [Google Scholar] [CrossRef] [PubMed]
- Pumroy, R.A.; Cingolani, G. Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem. J. 2015, 466, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, R.; Kinnunen, L.; Kohler, M.; Julkunen, I.; Melen, K. NF-κB is transported into the nucleus by importin α3 and importin α4. J. Biol. Chem. 2005, 280, 15942–15951. [Google Scholar] [CrossRef]
- Baker, S.A.; Lombardi, L.M.; Zoghbi, H.Y. Karyopherin α 3 and karyopherin α 4 proteins mediate the nuclear import of methyl-CpG binding protein 2. J. Biol. Chem. 2015, 290, 22485–22493. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, B.; Hartmann, H.; May, S.; Mohl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [CrossRef]
- Xu, S.; Xie, J.; Zhang, X.; Chen, L.; Bi, Y.; Li, X.; Idris, A.; Feng, R. DDX56 antagonizes IFN-β production to enhance EMCV replication by inhibiting IRF3 nuclear translocation. Vet. Microbiol. 2022, 264, 109304. [Google Scholar] [CrossRef]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; van-Minnen, J.; Twiss, J.L.; et al. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef]
- Chu, C.T.; Plowey, E.D.; Wang, Y.; Patel, V.; Jordan-Sciutto, K.L. Location, location, location: Altered transcription factor trafficking in neurodegeneration. J. Neuropathol. Exp. Neurol. 2007, 66, 873–883. [Google Scholar] [CrossRef]
- Ben-Yaakov, K.; Dagan, S.Y.; Segal-Ruder, Y.; Shalem, O.; Vuppalanchi, D.; Willis, D.E.; Yudin, D.; Rishal, I.; Rother, F.; Bader, M.; et al. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012, 31, 1350–1363. [Google Scholar] [CrossRef]
- Perry, R.B.; Doron-Mandel, E.; Iavnilovitch, E.; Rishal, I.; Dagan, S.Y.; Tsoory, M.; Coppola, G.; McDonald, M.K.; Gomes, C.; Geschwind, D.H.; et al. Subcellular knockout of importin beta1 perturbs axonal retrograde signaling. Neuron 2012, 75, 294–305. [Google Scholar] [CrossRef]
- Fainzilber, M.; Budnik, V.; Segal, R.A.; Kreutz, M.R. From synapse to nucleus and back again--communication over distance within neurons. J. Neurosci. 2011, 31, 16045–16048. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.R.; Otis, K.O.; Chen, D.Y.; Zhao, Y.; O’Dell, T.J.; Martin, K.C. Synapse to nucleus signaling during long-term synaptic plasticity; a role for the classical active nuclear import pathway. Neuron 2004, 44, 997–1009. [Google Scholar] [CrossRef]
- Panayotis, N.; Sheinin, A.; Dagan, S.Y.; Tsoory, M.M.; Rother, F.; Vadhvani, M.; Meshcheriakova, A.; Koley, S.; Marvaldi, L.; Song, D.A.; et al. Importin α5 Regulates Anxiety through MeCP2 and Sphingosine Kinase 1. Cell Rep. 2018, 25, 3169–3179.e7. [Google Scholar] [CrossRef]
- Terenzio, M.; Schiavo, G.; Fainzilber, M. Compartmentalized Signaling in Neurons: From Cell Biology to Neuroscience. Neuron 2017, 96, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Ramaswami, G.; Geschwind, D.H. Gene co-expression network analysis in human spinal cord highlights mechanisms underlying amyotrophic lateral sclerosis susceptibility. Sci. Rep. 2021, 11, 5748. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Chung, C.G.; Park, S.S.; Lee, D.; Kim, K.M.; Jeong, Y.; Kim, E.S.; Cho, J.H.; Jeon, Y.M.; Shen, C.J.; et al. Cytosolic calcium regulates cytoplasmic accumulation of TDP-43 through Calpain-A and Importin α3. Elife 2020, 9, e60132. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016, 132, 159–173. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovicic, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef]
- Zhang, J.; Ito, H.; Wate, R.; Ohnishi, S.; Nakano, S.; Kusaka, H. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006, 112, 673–680. [Google Scholar] [CrossRef]
- Schob, C.; Hempel, M.; Safka Brozkova, D.; Jiang, H.; Kim, S.Y.; Batzir, N.A.; Orenstein, N.; Bierhals, T.; Johannsen, J.; Uhrova Meszarosova, A.; et al. Dominant KPNA3 Mutations Cause Infantile-Onset Hereditary Spastic Paraplegia. Ann. Neurol. 2021, 90, 738–750. [Google Scholar] [CrossRef]
- Estiar, M.A.; Lail, N.; Dyment, D.A.; Varghaei, P.; Hartley, T.; Gillespie, M.K.; Yoon, G.; Boycott, K.M.; Rouleau, G.A.; Gan-Or, Z. Heterozygous De Novo KPNA3 Mutations Cause Complex Hereditary Spastic Paraplegia. Ann. Neurol. 2022, 91, 730–732. [Google Scholar] [CrossRef] [PubMed]
- De Winter, J.; Van de Vondel, L.; Zuchner, S.; Ortibus, E.; Baets, J. A Recurrent KPNA3 Missense Variant Causing Infantile Pure Spastic Paraplegia. Ann. Neurol. 2022, 91, 298–299. [Google Scholar] [CrossRef]
- Marvaldi, L.; Panayotis, N.; Alber, S.; Dagan, S.Y.; Okladnikov, N.; Koppel, I.; Di Pizio, A.; Song, D.A.; Tzur, Y.; Terenzio, M.; et al. Importin α3 regulates chronic pain pathways in peripheral sensory neurons. Science 2020, 369, 842–846. [Google Scholar] [CrossRef]
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 2018, 14, e1007363. [Google Scholar] [CrossRef] [PubMed]
- Renvoise, B.; Stadler, J.; Singh, R.; Bakowska, J.C.; Blackstone, C. Spg20-/- mice reveal multimodal functions for Troyer syndrome protein spartin in lipid droplet maintenance, cytokinesis and BMP signaling. Hum. Mol. Genet. 2012, 21, 3604–3618. [Google Scholar] [CrossRef]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef]
- Rother, F.; Shmidt, T.; Popova, E.; Krivokharchenko, A.; Hugel, S.; Vilianovich, L.; Ridders, M.; Tenner, K.; Alenina, N.; Kohler, M.; et al. Importin α7 is essential for zygotic genome activation and early mouse development. PLoS ONE 2011, 6, e18310. [Google Scholar] [CrossRef] [PubMed]
- Thiele, S.; Stanelle-Bertram, S.; Beck, S.; Kouassi, N.M.; Zickler, M.; Muller, M.; Tuku, B.; Resa-Infante, P.; van Riel, D.; Alawi, M.; et al. Cellular Importin-α3 Expression Dynamics in the Lung Regulate Antiviral Response Pathways against Influenza A Virus Infection. Cell Rep. 2020, 31, 107549. [Google Scholar] [CrossRef]
- Guyenet, S.J.; Furrer, S.A.; Damian, V.M.; Baughan, T.D.; La Spada, A.R.; Garden, G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010, 39, e1787. [Google Scholar] [CrossRef]
- Ferreira, P.A. The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell. Mol. Life Sci. 2019, 76, 2247–2273. [Google Scholar] [CrossRef]
- Basu, S.; Rajendra, K.C.; Alagar, S.; Bahadur, R.P. Impaired nuclear transport induced by juvenile ALS causing P525L mutation in NLS domain of FUS: A molecular mechanistic study. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140766. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Teramoto, S.; Kwak, S. Phosphorylated TDP-43 becomes resistant to cleavage by calpain: A regulatory role for phosphorylation in TDP-43 pathology of ALS/FTLD. Neurosci. Res. 2016, 107, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Plessis-Belair, J.; Ravano, K.; Han, E.; Janniello, A.; Molina, C.; Sher, R.B. NEMF mutations in mice illustrate how Importin-β specific nuclear transport defects recapitulate neurodegenerative disease hallmarks. PLoS Genet. 2024, 20, e1011411. [Google Scholar] [CrossRef]
- Sowa, A.S.; Martin, E.; Martins, I.M.; Schmidt, J.; Depping, R.; Weber, J.J.; Rother, F.; Hartmann, E.; Bader, M.; Riess, O.; et al. Karyopherin α-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc. Natl. Acad. Sci. USA 2018, 115, E2624–E2633. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Khalil, B.; Chhangani, D.; Wren, M.C.; Smith, C.L.; Lee, J.H.; Li, X.; Puttinger, C.; Tsai, C.W.; Fortin, G.; Morderer, D.; et al. Nuclear import receptors are recruited by FG-nucleoporins to rescue hallmarks of TDP-43 proteinopathy. Mol. Neurodegener. 2022, 17, 80. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, J.; Henderson, M.J.; Yang, P.; Hagen, B.M.; Siddique, T.; Vogel, B.E.; Deng, H.X.; Fang, S. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. Elife 2017, 6, e23759. [Google Scholar] [CrossRef]
- Mergy, M.A.; Gowrishankar, R.; Davis, G.L.; Jessen, T.N.; Wright, J.; Stanwood, G.D.; Hahn, M.K.; Blakely, R.D. Genetic targeting of the amphetamine and methylphenidate-sensitive dopamine transporter: On the path to an animal model of attention-deficit hyperactivity disorder. Neurochem. Int. 2014, 73, 56–70. [Google Scholar] [CrossRef]
- Borbelyova, V.; Janisova, K.; Myslivecek, J.; Riljak, V. Sex-related differences in locomotion and climbing of C57Bl/6NTac mice in a novel environment. Physiol. Res. 2019, 68, S353–S359. [Google Scholar] [CrossRef]
- Tsao, C.H.; Wu, K.Y.; Su, N.C.; Edwards, A.; Huang, G.J. The influence of sex difference on behavior and adult hippocampal neurogenesis in C57BL/6 mice. Sci. Rep. 2023, 13, 17297. [Google Scholar] [CrossRef]
- Sakurai, K.; Morita, M.; Aomine, Y.; Matsumoto, M.; Moriyama, T.; Kasahara, E.; Sekiyama, A.; Otani, M.; Oshima, R.; Loveland, K.L.; et al. Importin α4 deficiency induces psychiatric disorder-related behavioral deficits and neuroinflammation in mice. Transl. Psychiatry 2024, 14, 426. [Google Scholar] [CrossRef] [PubMed]
- Faraone, S.V.; Bellgrove, M.A.; Brikell, I.; Cortese, S.; Hartman, C.A.; Hollis, C.; Newcorn, J.H.; Philipsen, A.; Polanczyk, G.V.; Rubia, K.; et al. Attention-deficit/hyperactivity disorder. Nat. Rev. Dis. Primers 2024, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Rovira, P.; Demontis, D.; Sanchez-Mora, C.; Zayats, T.; Klein, M.; Mota, N.R.; Weber, H.; Garcia-Martinez, I.; Pagerols, M.; Vilar-Ribo, L.; et al. Shared genetic background between children and adults with attention deficit/hyperactivity disorder. Neuropsychopharmacology 2020, 45, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Staller, J.; Faraone, S.V. Attention-deficit hyperactivity disorder in girls: Epidemiology and management. CNS Drugs 2006, 20, 107–123. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Hindlimb Clasping (per Number of Mice in Group) | p-Value | |
|---|---|---|
| Kpna3-WT females | 0/11 | >0.9999 |
| Kpna3-KO females | 1/11 | |
| Kpna3-WT males | 0/11 | >0.9999 |
| Kpna3-KO males | 0/11 | |
| Kpna4-WT females | 0/11 | 0.4762 |
| Kpna4-KO females | 1/10 | |
| Kpna4-WT males | 0/11 | >0.9999 |
| Kpna4-KO males | 1/11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rother, F.; Parmar, A.R.; Bodenhagen, J.S.; Marvaldi, L.; Hartmann, E.; Bader, M. Deficiency in KPNA4, but Not in KPNA3, Causes Attention Deficit/Hyperactivity Disorder like Symptoms in Mice. Genes 2025, 16, 690. https://doi.org/10.3390/genes16060690
Rother F, Parmar AR, Bodenhagen JS, Marvaldi L, Hartmann E, Bader M. Deficiency in KPNA4, but Not in KPNA3, Causes Attention Deficit/Hyperactivity Disorder like Symptoms in Mice. Genes. 2025; 16(6):690. https://doi.org/10.3390/genes16060690
Chicago/Turabian StyleRother, Franziska, Amishaben R. Parmar, Julia S. Bodenhagen, Letizia Marvaldi, Enno Hartmann, and Michael Bader. 2025. "Deficiency in KPNA4, but Not in KPNA3, Causes Attention Deficit/Hyperactivity Disorder like Symptoms in Mice" Genes 16, no. 6: 690. https://doi.org/10.3390/genes16060690
APA StyleRother, F., Parmar, A. R., Bodenhagen, J. S., Marvaldi, L., Hartmann, E., & Bader, M. (2025). Deficiency in KPNA4, but Not in KPNA3, Causes Attention Deficit/Hyperactivity Disorder like Symptoms in Mice. Genes, 16(6), 690. https://doi.org/10.3390/genes16060690