
Systemic EBV+ T-Cell Lymphoma of Childhood with Hemophagocytic Lymphohistiocytosis in a Patient with a Highly Complex Karyotype


Abstract
1. Introduction
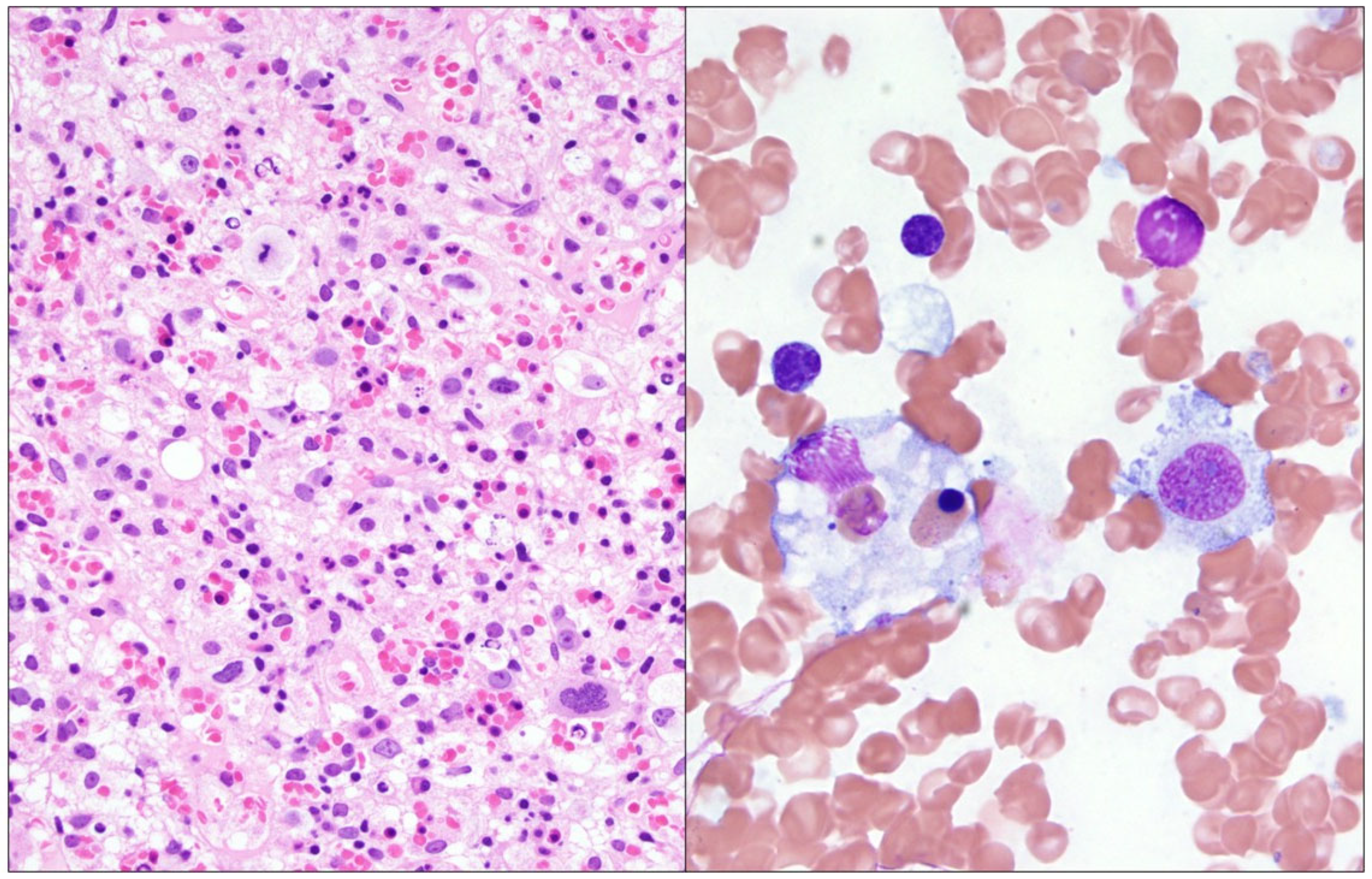
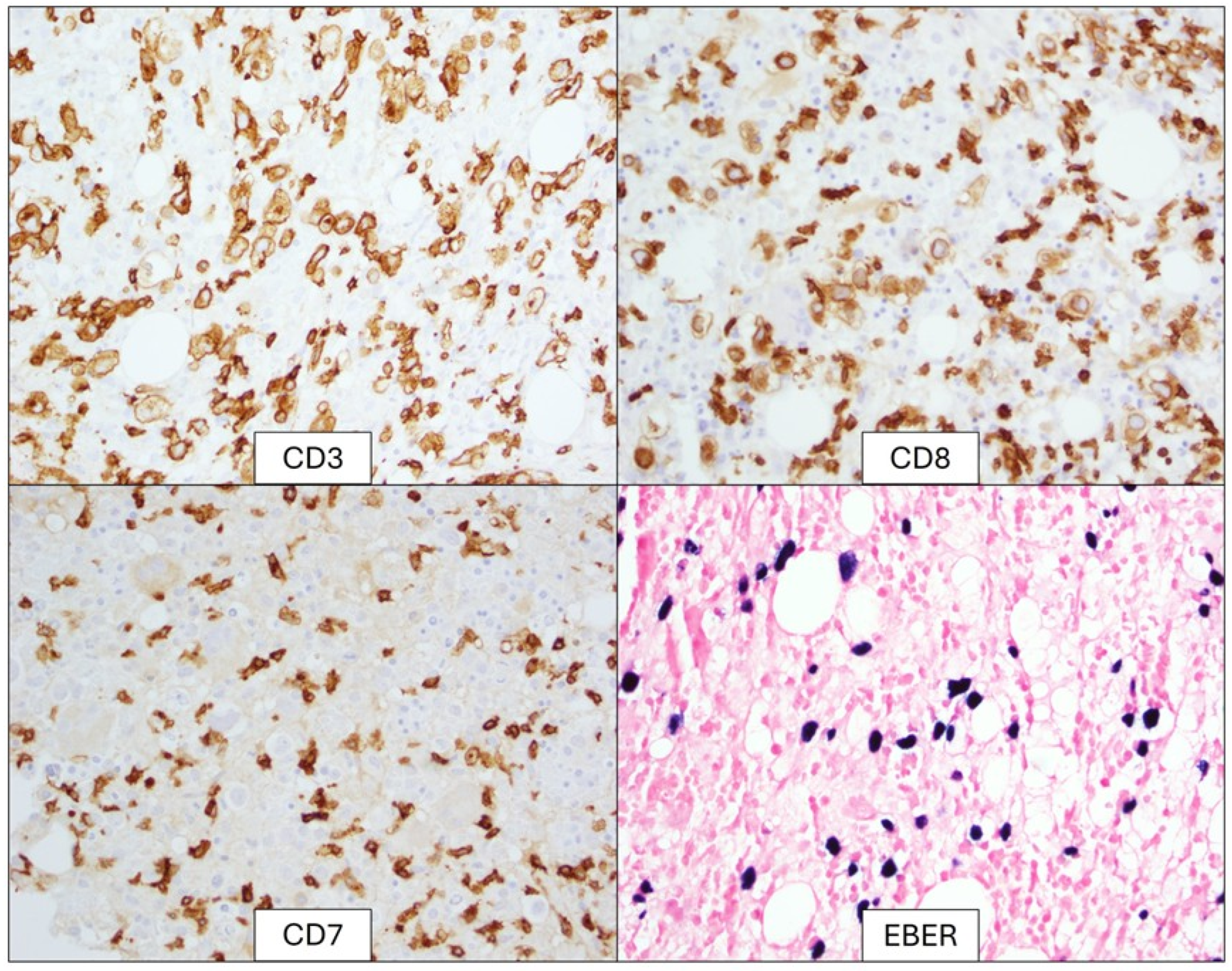
2. Detailed Case Description
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deng, W.; Xu, Y.; Yuan, X. Clinical features and prognosis of acute lymphoblastic leukemia in children with Epstein-Barr virus infection. Transl. Pediatr. 2022, 11, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Baumforth, K.R.; Young, L.S.; Flavell, K.J.; Constandinou, C.; Murray, P.G. The Epstein-Barr virus and its association with human cancers. Mol. Pathol. 1999, 52, 307–322. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quintanilla-Martinez, L.; Kimura, H.; Jaffe, E. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues; Swerdlow, S., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J., Eds.; IARCL: Lyon, France, 2008; EBV+ T-cell Lymphoproliferative Disorders of Childhood; pp. 278–280. [Google Scholar]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Swerdlow, S.H.; Tousseyn, T.; Barrionuevo, C.; Nakamura, S.; Jaffe, E.S. New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders. Virchows Arch. 2023, 482, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Standage, S.W.; Filipovich, A.H. Hemophagocytic Lymphohistiocytosis Syndromes. Pediatr. Crit. Care Med. 2014, 385–393. [Google Scholar] [CrossRef] [PubMed Central]
- Zhang, K.; Astigarraga, I.; Bryceson, Y.; Lehmberg, K.; Machowicz, R.; Marsh, R.; Sieni, E.; Wang, Z.; Nichols, K.E. Familial Hemophagocytic Lymphohistiocytosis. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2006. [Google Scholar]
- Konkol, S.; Rai, M. Lymphohistiocytosis. [Updated 2023 Mar 27]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557776/ (accessed on 4 January 2025).
- Kim, W.Y.; Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. Epstein-Barr Virus-Associated T and NK-Cell Lymphoproliferative Diseases. Front. Pediatr. 2019, 7, 71. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jimenez, A.; Carrillo, L.F.; Carlsen, E.D. Systemic EBV-positive T-cell lymphoma of childhood with unusual cytogenetic findings and family history of Still’s disease-associated recurrent macrophage activation syndrome. Pediatr. Blood Cancer 2024, 71, e31291. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Cohen, D.N.; Greig, B.; Yenamandra, A.; Vnencak-Jones, C.; Thompson, M.A.; Kim, A.S. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int. J. Clin. Exp. Pathol. 2014, 7, 5738–5749. [Google Scholar] [PubMed] [PubMed Central]
- Millot, F.; Brizard, F.; Babin, P.; Canioni, D.; Macintyre, E.; Levard, G.; Gambert, C.; Ricour, C.; Guilhot, F. t(3;17)(q21;q25) in Epstein-Barr virus associated peripheral T-cell lymphoma: A paediatric case. Br. J. Haematol. 1998, 100, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.; Sreenivasan, S.; Peterson, C.; Rai, M.; Kancharla, P.; Santosa, S.; Bunker, M.; Samhouri, Y. Systemic Epstein-Barr Virus-Positive T-Cell Lymphoma of Childhood Associated with t(1;22)(p22;q11.2) Mutation. J. Hematol. 2024, 13, 229–237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mian, A.; Kumari, K.; Kaushal, S.; Fazal, F.; Kodan, P.; Batra, A.; Kumar, P.; Baitha, U.; Jorwal, P.; Soneja, M.; et al. Fatal familial hemophagocytic lymphohistiocytosis with perforin gene (PRF1) mutation and EBV-associated T-cell lymphoproliferative disorder of the thyroid. Autops. Case Rep. 2019, 9, e2019101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, J.; Wang, D.; Nassiri, M. Clonal CD8+ T Lymphocytic Proliferation and Karyotypical Abnormalities in an EBV Associated Hemophagocytic Lymphohistiocytosis. Case Rep. Pathol. 2015, 2015, 513968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zehr, B.; Brannock, K.; Wyma, R.; Kahwash, S.B. Differentiating fulminant EBV infection complicated by HLH from Lymphoma: Report of a case and a brief literature review. Diagn. Pathol. 2023, 18, 28. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ito, E.; Kitazawa, J.; Arai, K.; Otomo, H.; Endo, Y.; Imashuku, S.; Yokoyama, M. Fatal Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis with clonal karyotype abnormality. Int. J. Hematol. 2000, 71, 263–265. [Google Scholar] [PubMed]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) ISCN 2020 an International System for Human Cytogenomic Nomenclature; S. Karger Publishers: Basel, Switzerland, 2020. [Google Scholar]
- Henter, J.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, W.; Weisberger, J.; Liu, Z.; Tsang, P.; Hossein, M.; Wu, C.D.; Dong, H.; Wong, J.Y.; Tugulea, S.; Dee, S.; et al. An approach to diagnosis of T-cell lymphoproliferative disorders by flow cytometry. Cytometry 2002, 50, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Krishna, M.T.; Pickard, C.; Smith, J.L. Diagnostic role of tests for T cell receptor (TCR) genes. J. Clin. Pathol. 2003, 56, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Coffey, A.M.; Lewis, A.; Marcogliese, A.N.; Elghetany, M.T.; Punia, J.N.; Chang, C.; Allen, C.E.; McClain, K.L.; Gaikwad, A.S.; El-Mallawany, N.K.; et al. A clinicopathologic study of the spectrum of systemic forms of EBV-associated T-cell lymphoproliferative disorders of childhood: A single tertiary care pediatric institution experience in North America. Pediatr. Blood Cancer 2019, 66, e27798. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, S.; Silva, O.; Natkunam, Y. Additional considerations related to the elusive boundaries of EBV-associated T/NK-cell lymphoproliferative disorders. Haematologica 2019, 104, e125–e126. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Z.; Guan, P. Rethinking the elusive boundaries of EBV-associated T/NK-cell lymphoproliferative disorders. Haematologica 2019, 104, e124–e125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Finding/Value | Reference Range |
|---|---|---|
| Temperature | 39.4 °C | 36.1–37.2 °C |
| Hemoglobin | 8.0 g/dL | 12.0–16.0 g/dL |
| Platelet count | 53 × 103 µL | 150–400 × 103 µL |
| Absolute neutrophil count | 0.72 × 103 µL | 1.7–7.00 × 103 µL |
| Triglycerides | 608 mg/dL | <150 mg/dL |
| Fibrinogen | 166 mg/dL | 173–454 mg/dL |
| Ferritin | >13,000 ng/mL | 11–204 ng/mL |
| Splenomegaly | 22.7 cm | <12 cm |
| Soluble CD25 | 66,395.7 pg/mL | 175.3–858.2 pg/mL |
| CXCL9 | 89,046 pg/mL | <647 pg/mL |
| EBV Quant PCR | 218,658 IU/mL | Not detectable |
| Hemophagocytosis in marrow | Present | Absent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maher, P.; Guzman, E.; Chaffin, J.; Kashif, R.; Burnside, R.D. Systemic EBV+ T-Cell Lymphoma of Childhood with Hemophagocytic Lymphohistiocytosis in a Patient with a Highly Complex Karyotype. Genes 2025, 16, 382. https://doi.org/10.3390/genes16040382
Maher P, Guzman E, Chaffin J, Kashif R, Burnside RD. Systemic EBV+ T-Cell Lymphoma of Childhood with Hemophagocytic Lymphohistiocytosis in a Patient with a Highly Complex Karyotype. Genes. 2025; 16(4):382. https://doi.org/10.3390/genes16040382
Chicago/Turabian StyleMaher, Patrick, Emilia Guzman, Joanna Chaffin, Reema Kashif, and Rachel D. Burnside. 2025. "Systemic EBV+ T-Cell Lymphoma of Childhood with Hemophagocytic Lymphohistiocytosis in a Patient with a Highly Complex Karyotype" Genes 16, no. 4: 382. https://doi.org/10.3390/genes16040382
APA StyleMaher, P., Guzman, E., Chaffin, J., Kashif, R., & Burnside, R. D. (2025). Systemic EBV+ T-Cell Lymphoma of Childhood with Hemophagocytic Lymphohistiocytosis in a Patient with a Highly Complex Karyotype. Genes, 16(4), 382. https://doi.org/10.3390/genes16040382