
Cardiac Fibroblasts: Helping or Hurting

 , ,
, ,
Abstract
1. Introduction
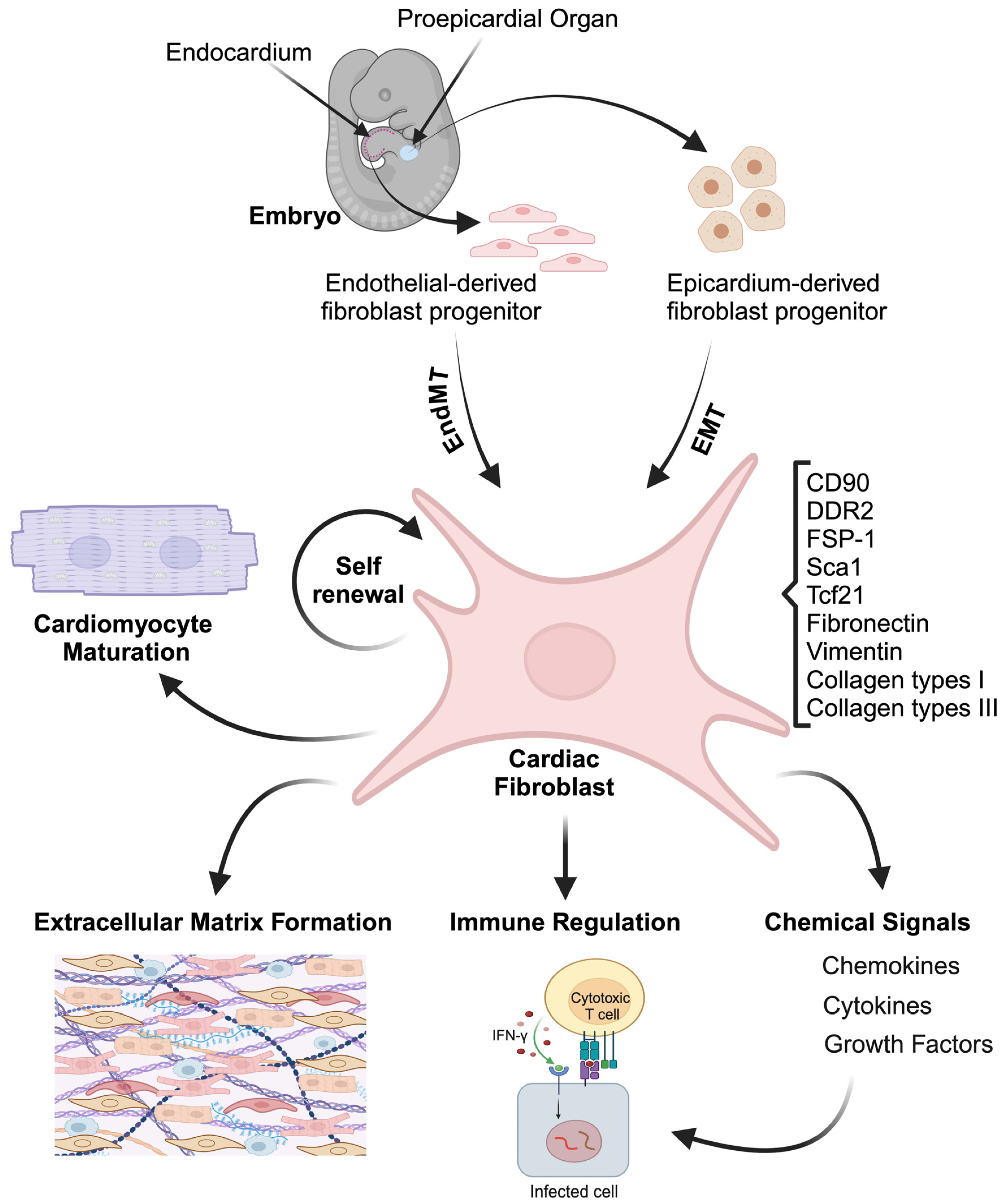
2. Cardiac Fibroblasts: Origin and Function
2.1. Cardiac Fibroblasts Origin
2.2. Cardiac Fibroblasts Function
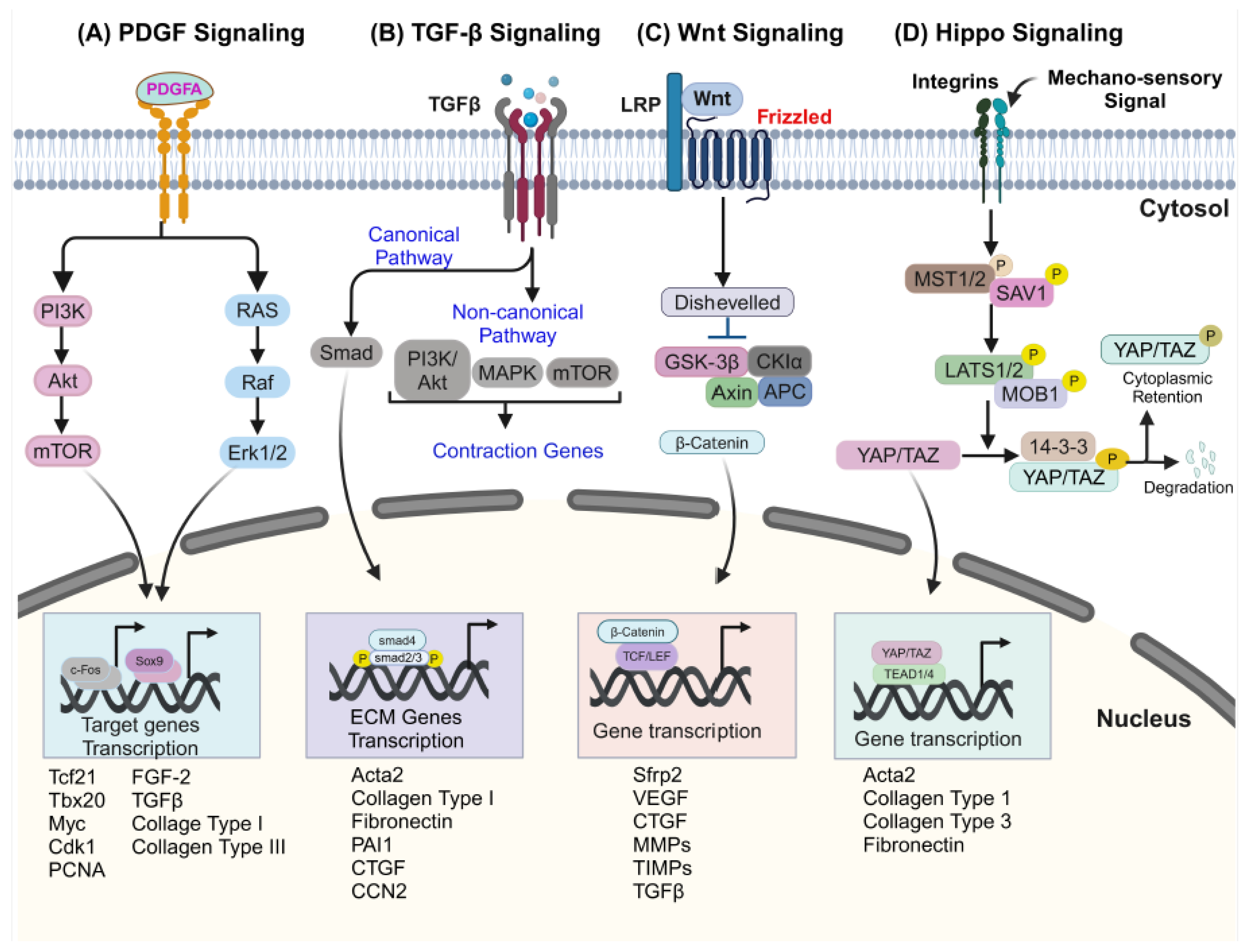
3. Signaling in CF During Cardiac Pathogenesis
3.1. Platelet-Derived Growth Factor Receptors (PDGFRs) Signaling in CFs
3.2. Transforming Growth Factor-β (TGF-β) Signaling in CFs
3.3. Wingless-Related Integration Site (Wnt) Signaling in CFs
3.4. Hippo Signaling in CFs
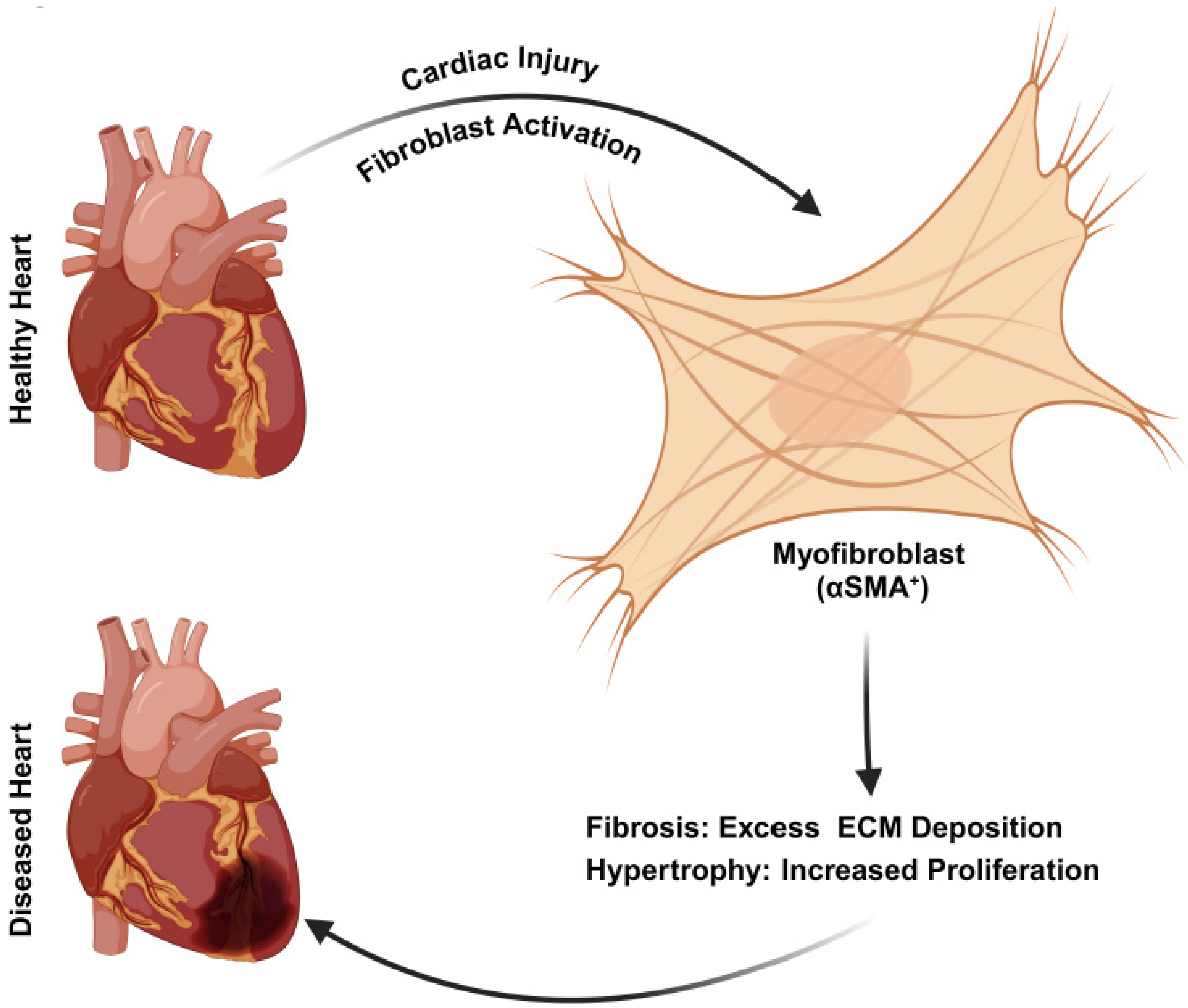
4. Cardiac Fibroblasts in Cardiovascular Diseases
4.1. Cardiac Fibroblasts in Hypertrophic Cardiomyopathy
4.2. Cardiac Fibroblasts in Dilated Cardiomyopathy
4.3. Cardiac Fibroblasts in Muscular Dystrophy
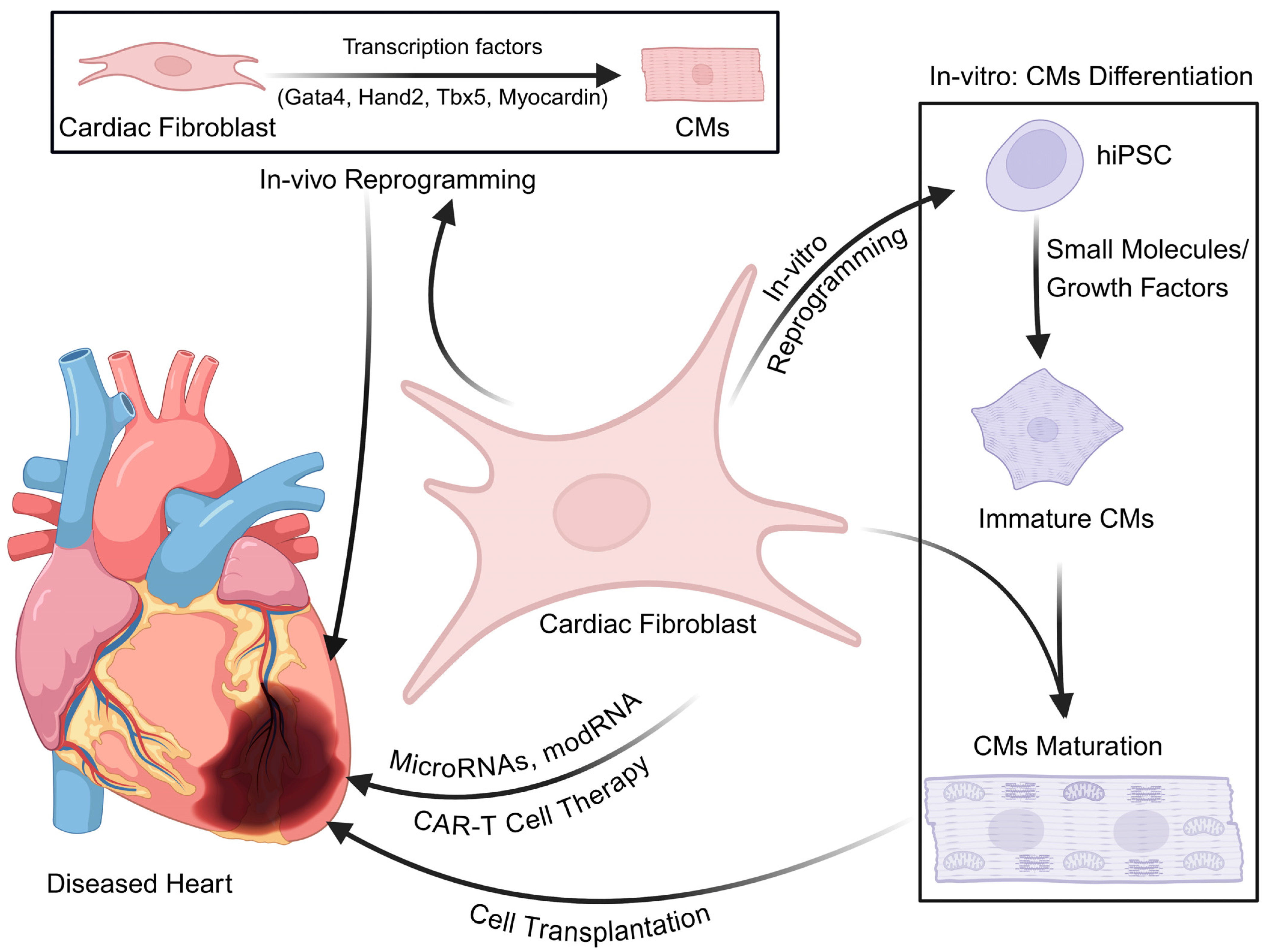
5. Strategies to Mitigate Cardiac Fibrosis and Therapeutics
5.1. Pharmalogical Interventions for Cardiac Diseases
5.2. Cell–Gene-Based Therapeutic Approach for Cardiac Diseases
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.N.; Manuel, F.; Nascimento, D.S. The bright side of fibroblasts: Molecular signature and regenerative cues in major organs. NPJ Regen. Med. 2021, 6, 43. [Google Scholar] [CrossRef]
- Abercrombie, M.; Flint, M.H.; James, D.W. Wound Contraction in Relation to Collagen Formation in Scorbutic Guinea-Pigs. J. Embryol. Exp. Morphol. 1956, 4, 167–175. [Google Scholar]
- Abercrombie, M.; Heaysman, J.E.; Pegrum, S.M. The locomotion of fibroblasts in culture. 3. Movements of particles on the dorsal surface of the leading lamella. Exp. Cell Res. 1970, 62, 389–398. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.B.; Majno, G. Presence of Modified Fibroblasts in Granulation Tissue and Their Possible Role in Wound Contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Carrel, A. On the permanent life of tissues outside of the organism. J. Exp. Med. 1912, 15, U516–U530. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative Studies of Growth of Mouse Embryo Cells in Culture and Their Development into Established Lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Herriges, M.; Morrisey, E.E. Lung development: Orchestrating the generation and regeneration of a complex organ. Development 2014, 141, 502–513. [Google Scholar] [CrossRef]
- Lendahl, U.; Muhl, L.; Betsholtz, C. Identification, discrimination and heterogeneity of fibroblasts. Nat. Commun. 2022, 13, 3409. [Google Scholar] [CrossRef]
- Mikawa, T.; Gourdie, R.G. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 1996, 174, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Serluca, F.C. Development of the proepicardial organ in the zebrafish. Dev. Biol. 2008, 315, 18–27. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef]
- Han, M.; Liu, Z.; Liu, L.; Huang, X.; Wang, H.; Pu, W.; Wang, E.; Liu, X.; Li, Y.; He, L.; et al. Dual genetic tracing reveals a unique fibroblast subpopulation modulating cardiac fibrosis. Nat. Genet. 2023, 55, 665–678. [Google Scholar] [CrossRef]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef]
- Fernandes, I.; Funakoshi, S.; Hamidzada, H.; Epelman, S.; Keller, G. Modeling cardiac fibroblast heterogeneity from human pluripotent stem cell-derived epicardial cells. Nat. Commun. 2023, 14, 8183. [Google Scholar] [CrossRef]
- Goldsmith, E.C.; Hoffman, A.; Morales, M.O.; Potts, J.D.; Price, R.L.; McFadden, A.; Rice, M.; Borg, T.K. Organization of fibroblasts in the heart. Dev. Dyn. 2004, 230, 787–794. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.N.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Cleutjens, J.P.M.; Kandala, J.C.; Guarda, E.; Guntaka, R.V.; Weber, K.T. Regulation of Collagen Degradation in the Rat Myocardium after Infarction. J. Mol. Cell. Cardiol. 1995, 27, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef]
- Furtado, M.B.; Costa, M.W.; Pranoto, E.A.; Salimova, E.; Pinto, A.R.; Lam, N.T.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.P.; et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef]
- Tang, Y.; Aryal, S.; Geng, X.; Zhou, X.; Fast, V.G.; Zhang, J.; Lu, R.; Zhou, Y. TBX20 Improves Contractility and Mitochondrial Function During Direct Human Cardiac Reprogramming. Circulation 2022, 146, 1518–1536. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef]
- Tolonen, J.P.; Salo, A.M.; Finnila, M.; Aro, E.; Karjalainen, E.; Ronkainen, V.P.; Drushinin, K.; Merceron, C.; Izzi, V.; Schipani, E.; et al. Reduced Bone Mass in Collagen Prolyl 4-Hydroxylase P4ha1 (+/−); P4ha2 (−/−) Compound Mutant Mice. JBMR Plus 2022, 6, e10630. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Zhang, D.; Zhou, X.; Xie, J. Role of the fibroblast growth factor 19 in the skeletal system. Life Sci. 2021, 265, 118804. [Google Scholar] [CrossRef]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Sc, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Stouffs, M.; Bettiol, E.; Kampf, S.; Krause, K.H.; Chaponnier, C.; Jaconi, M. Expression and function of alpha-smooth muscle actin during embryonic-stem-cell-derived cardiomyocyte differentiation. J. Cell Sci. 2007, 120, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Gu, Y.; Li, P.; Johnson, B.L.; Sucov, H.M.; Thomas, P.S. PDGF-A as an epicardial mitogen during heart development. Dev. Dyn. 2008, 237, 692–701. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Cattaneo, P.; Puceat, M.; Evans, S.M. Origins of cardiac fibroblasts. J. Mol. Cell Cardiol. 2016, 91, 1–5. [Google Scholar] [CrossRef]
- Oliveira, C.B.; Romo-Tena, J.; Patino-Martinez, E.; Woo, A.; Byrd, A.S.; Kim, D.; Okoye, G.A.; Kaplan, M.J.; Carmona-Rivera, C. Neutrophil extracellular traps activate Notch-gamma-secretase signaling in hidradenitis suppurativa. J. Allergy Clin. Immunol. 2025, 155, 188–198. [Google Scholar] [CrossRef]
- Valente, M.; Nascimento, D.S.; Cumano, A.; Pinto-do, O.P. Sca-1+ cardiac progenitor cells and heart-making: A critical synopsis. Stem Cells Dev. 2014, 23, 2263–2273. [Google Scholar] [CrossRef]
- Preston, K.J.; Kawai, T.; Torimoto, K.; Kuroda, R.; Nakayama, Y.; Akiyama, T.; Kimura, Y.; Scalia, R.; Autieri, M.V.; Rizzo, V.; et al. Mitochondrial fission inhibition protects against hypertension induced by angiotensin II. Hypertens. Res. 2024, 47, 1338–1349. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, S.; Ishii, Y.; Hamashima, T.; Yamamoto, S.; Mori, H.; Fujimori, T.; Shen, J.; Inoue, R.; Nishizono, H.; Itoh, H.; et al. PDGFRalpha plays a crucial role in connective tissue remodeling. Sci. Rep. 2015, 5, 17948. [Google Scholar] [CrossRef]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.J.; Kuwabara, J.T.; Riggsbee, K.L.; Tallquist, M.D. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H330–H344. [Google Scholar] [CrossRef]
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.T.; Shaw, R.M.; Srivastava, D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 2009, 16, 233–244. [Google Scholar] [CrossRef]
- Johnson, R.D.; Camelliti, P. Role of Non-Myocyte Gap Junctions and Connexin Hemichannels in Cardiovascular Health and Disease: Novel Therapeutic Targets? Int. J. Mol. Sci. 2018, 19, 866. [Google Scholar] [CrossRef]
- Kuwabara, J.T.; Hara, A.; Bhutada, S.; Gojanovich, G.S.; Chen, J.; Hokutan, K.; Shettigar, V.; Lee, A.Y.; DeAngelo, L.P.; Heckl, J.R.; et al. Consequences of PDGFRalpha(+) fibroblast reduction in adult murine hearts. Elife 2022, 11, e69854. [Google Scholar] [CrossRef]
- Hortells, L.; Valiente-Alandi, I.; Thomas, Z.M.; Agnew, E.J.; Schnell, D.J.; York, A.J.; Vagnozzi, R.J.; Meyer, E.C.; Molkentin, J.D.; Yutzey, K.E. A specialized population of Periostin-expressing cardiac fibroblasts contributes to postnatal cardiomyocyte maturation and innervation. Proc. Natl. Acad. Sci. USA 2020, 117, 21469–21479. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, F.; Wang, L.; Li, Z.; Ren, Z.; Li, D.; Zhang, M.; Han, L.; Wang, S.Q.; Zhou, B.; et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 2020, 11, 2585. [Google Scholar] [CrossRef]
- Ebeid, D.E.; Khalafalla, F.G.; Broughton, K.M.; Monsanto, M.M.; Esquer, C.Y.; Sacchi, V.; Hariharan, N.; Korski, K.I.; Moshref, M.; Emathinger, J.; et al. Pim1 maintains telomere length in mouse cardiomyocytes by inhibiting TGFbeta signalling. Cardiovasc. Res. 2021, 117, 201–211. [Google Scholar] [CrossRef]
- Singh, B.N.; Yucel, D.; Garay, B.I.; Tolkacheva, E.G.; Kyba, M.; Perlingeiro, R.C.R.; van Berlo, J.H.; Ogle, B.M. Proliferation and Maturation: Janus and the Art of Cardiac Tissue Engineering. Circ. Res. 2023, 132, 519–540. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.; van Veen, T.A.; van Rijen, H.V.; de Bakker, J.M. Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharmacol. 2011, 57, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Valiente-Alandi, I.; Nieman, M.L.; Sargent, M.A.; Lorenz, J.N.; Molkentin, J.D.; Blaxall, B.C. Pharmacological and Activated Fibroblast Targeting of Gbetagamma-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression. J. Am. Coll. Cardiol. 2017, 70, 958–971. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Tenkorang, M.A.A.; Chalise, U.; Konfrst, S.R.; Lindsey, M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020, 91–92, 109–116. [Google Scholar] [CrossRef]
- Lim, W.Y.; Lloyd, G.; Bhattacharyya, S. Mechanical and surgical bioprosthetic valve thrombosis. Heart 2017, 103, 1934–1941. [Google Scholar] [CrossRef]
- He, Z.Q.; Yuan, X.W.; Lu, Z.B.; Li, Y.H.; Li, Y.F.; Liu, X.; Wang, L.; Zhang, Y.; Zhou, Q.; Li, W. Pharmacological regulation of tissue fibrosis by targeting the mechanical contraction of myofibroblasts. Fundam. Res. 2022, 2, 37–47. [Google Scholar] [CrossRef]
- Noskovicova, N.; Hinz, B.; Pakshir, P. Implant Fibrosis and the Underappreciated Role of Myofibroblasts in the Foreign Body Reaction. Cells 2021, 10, 1794. [Google Scholar] [CrossRef]
- Cartledge, J.E.; Kane, C.; Dias, P.; Tesfom, M.; Clarke, L.; McKee, B.; Al Ayoubi, S.; Chester, A.; Yacoub, M.H.; Camelliti, P.; et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc. Res. 2015, 105, 260–270. [Google Scholar] [CrossRef]
- Ottaviano, F.G.; Yee, K.O. Communication signals between cardiac fibroblasts and cardiac myocytes. J. Cardiovasc. Pharmacol. 2011, 57, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhao, W.; Chen, Y.; Li, V.S.; Meng, W.; Sun, Y. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1719–H1726. [Google Scholar] [CrossRef] [PubMed]
- Bergsten, E.; Uutela, M.; Li, X.; Pietras, K.; Ostman, A.; Heldin, C.H.; Alitalo, K.; Eriksson, U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 2001, 3, 512–516. [Google Scholar] [CrossRef]
- Hamid, T.; Xu, Y.; Ismahil, M.A.; Rokosh, G.; Jinno, M.; Zhou, G.; Wang, Q.; Prabhu, S.D. Cardiac Mesenchymal Stem Cells Promote Fibrosis and Remodeling in Heart Failure: Role of PDGF Signaling. JACC Basic. Transl. Sci. 2022, 7, 465–483. [Google Scholar] [CrossRef]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef]
- Ponten, A.; Li, X.; Thoren, P.; Aase, K.; Sjoblom, T.; Ostman, A.; Eriksson, U. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. Am. J. Pathol. 2003, 163, 673–682. [Google Scholar] [CrossRef]
- Ponten, A.; Folestad, E.B.; Pietras, K.; Eriksson, U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ. Res. 2005, 97, 1036–1045. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Zymek, P.; Bujak, M.; Chatila, K.; Cieslak, A.; Thakker, G.; Entman, M.L.; Frangogiannis, N.G. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J. Am. Coll. Cardiol. 2006, 48, 2315–2323. [Google Scholar] [CrossRef]
- Hume, R.D.; Deshmukh, T.; Doan, T.; Shim, W.J.; Kanagalingam, S.; Tallapragada, V.; Rashid, F.; Marcuello, M.; Blessing, D.; Selvakumar, D.; et al. PDGF-AB Reduces Myofibroblast Differentiation Without Increasing Proliferation After Myocardial Infarction. JACC Basic. Transl. Sci. 2023, 8, 658–674. [Google Scholar] [CrossRef]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, C.K.; Robinson, E.L.; Abdesselem, M.; Trenson, S.; Dries, E.; Gilbert, G.; Janssens, S.; Van Cleemput, J.; Rega, F.; Meyns, B.; et al. Myofibroblast Phenotype and Reversibility of Fibrosis in Patients With End-Stage Heart Failure. J. Am. Coll. Cardiol. 2019, 73, 2267–2282. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Massague, J.; Sheppard, D. TGF-beta signaling in health and disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef]
- Yuan, W.; Varga, J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J. Biol. Chem. 2001, 276, 38502–38510. [Google Scholar] [CrossRef]
- Leask, A.; Holmes, A.; Black, C.M.; Abraham, D.J. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J. Biol. Chem. 2003, 278, 13008–13015. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Jin, J.; Shen, W.; Tsui, H.; Shi, Y.; Wang, Y.; Zhang, Y.; Hao, G.; Wu, J.; Chen, S.; et al. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J. Am. Heart Assoc. 2014, 3, e000340. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Z.; Morsink, M.A.; Kim, S.W.; Luo, L.J.; Zhang, X.; Soni, R.K.; Lock, R.I.; Rao, J.; Kim, Y.; Zhang, A.; et al. Cardiac fibroblast BAG3 regulates TGFBR2 signaling and fibrosis in dilated cardiomyopathy. J. Clin. Investig. 2025, 135, e181630. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Sokol, S.Y. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 2011, 138, 4341–4350. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Kobayashi, K.; Luo, M.; Zhang, Y.; Wilkes, D.C.; Ge, G.; Grieskamp, T.; Yamada, C.; Liu, T.C.; Huang, G.; Basson, C.T.; et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat. Cell Biol. 2009, 11, 46–55. [Google Scholar] [CrossRef]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef]
- Wu, B.; Crampton, S.P.; Hughes, C.C. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity 2007, 26, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Egea, V.; Zahler, S.; Rieth, N.; Neth, P.; Popp, T.; Kehe, K.; Jochum, M.; Ries, C. Tissue inhibitor of metalloproteinase-1 (TIMP-1) regulates mesenchymal stem cells through let-7f microRNA and Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E309–E316. [Google Scholar] [CrossRef] [PubMed]
- Dzialo, E.; Czepiel, M.; Tkacz, K.; Siedlar, M.; Kania, G.; Blyszczuk, P. WNT/beta-Catenin Signaling Promotes TGF-beta-Mediated Activation of Human Cardiac Fibroblasts by Enhancing IL-11 Production. Int. J. Mol. Sci. 2021, 22, 10072. [Google Scholar] [CrossRef]
- Xiang, F.L.; Fang, M.; Yutzey, K.E. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef]
- Cao, J.; Tsenovoy, P.L.; Thompson, E.A.; Falck, J.R.; Touchon, R.; Sodhi, K.; Rezzani, R.; Shapiro, J.I.; Abraham, N.G. Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat. 2015, 116–117, 76–86. [Google Scholar] [CrossRef]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef]
- Tanjore, H.; Degryse, A.L.; Crossno, P.F.; Xu, X.C.; McConaha, M.E.; Jones, B.R.; Polosukhin, V.V.; Bryant, A.J.; Cheng, D.S.; Newcomb, D.C.; et al. beta-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am. J. Respir. Crit. Care Med. 2013, 187, 630–639. [Google Scholar] [CrossRef]
- Carthy, J.M.; Garmaroudi, F.S.; Luo, Z.; McManus, B.M. Wnt3a induces myofibroblast differentiation by upregulating TGF-beta signaling through SMAD2 in a beta-catenin-dependent manner. PLoS ONE 2011, 6, e19809. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef]
- Song, S.; Zhang, X.; Huang, Z.; Zhao, Y.; Lu, S.; Zeng, L.; Cai, F.; Wang, T.; Pei, Z.; Weng, X.; et al. TEA domain transcription factor 1(TEAD1) induces cardiac fibroblasts cells remodeling through BRD4/Wnt4 pathway. Signal Transduct. Target. Ther. 2024, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef]
- Lin, Z.; Zhou, P.; von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; van Gorp, P.R.; Wang, D.Z.; Pu, W.T. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Badouel, C.; McNeill, H. SnapShot: The hippo signaling pathway. Cell 2011, 145, 484.e1. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Y.; Ai, D.; Yao, L.; Jiang, H. The role of the Hippo pathway in heart disease. FEBS J. 2022, 289, 5819–5833. [Google Scholar] [CrossRef]
- Wang, P.; Mao, B.; Luo, W.; Wei, B.; Jiang, W.; Liu, D.; Song, L.; Ji, G.; Yang, Z.; Lai, Y.Q.; et al. The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic. Res. Cardiol. 2014, 109, 435. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Cibi, D.M.; Ghani, S.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of YAP/TAZ in cardiac fibroblasts attenuates adverse remodelling and improves cardiac function. Cardiovasc. Res. 2022, 118, 1785–1804. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Ju, X.; Sun, H.; Finnerty, C.C.; Herndon, D.N.; Brasier, A.R. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J. Investig. Dermatol. 2013, 133, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Yekkala, K.; Borg, T.K.; Baudino, T.A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N. Y. Acad. Sci. 2006, 1080, 76–84. [Google Scholar] [CrossRef]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Thomas, T.P.; Grisanti, L.A. The Dynamic Interplay Between Cardiac Inflammation and Fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef]
- Fredj, S.; Bescond, J.; Louault, C.; Potreau, D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J. Cell Physiol. 2005, 202, 891–899. [Google Scholar] [CrossRef]
- van Wamel, A.J.; Ruwhof, C.; van der Valk-Kokshoom, L.E.; Schrier, P.I.; van der Laarse, A. The role of angiotensin II, endothelin-1 and transforming growth factor-beta as autocrine/paracrine mediators of stretch-induced cardiomyocyte hypertrophy. Mol. Cell Biochem. 2001, 218, 113–124. [Google Scholar] [CrossRef]
- Schlittler, M.; Pramstaller, P.P.; Rossini, A.; De Bortoli, M. Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Perspective from Fibroblasts. Int. J. Mol. Sci. 2023, 24, 14845. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ. Res. 2021, 128, 1533–1553. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Bogomolovas, J.; Chen, J. Understanding the molecular basis of cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H181–H233. [Google Scholar] [CrossRef] [PubMed]
- Golubenko, M.V.; Pavlyukova, E.N.; Salakhov, R.R.; Makeeva, O.A.; Puzyrev, K.V.; Glotov, O.S.; Puzyrev, V.P.; Nazarenko, M.S. A New Leu714Arg Variant in the Converter Domain of MYH7 is Associated with a Severe form of Familial Hypertrophic Cardiomyopathy. Front. Biosci. 2024, 16, 1. [Google Scholar] [CrossRef]
- Hsieh, J.; Becklin, K.L.; Givens, S.; Komosa, E.R.; Llorens, J.E.A.; Kamdar, F.; Moriarity, B.S.; Webber, B.R.; Singh, B.N.; Ogle, B.M. Myosin Heavy Chain Converter Domain Mutations Drive Early-Stage Changes in Extracellular Matrix Dynamics in Hypertrophic Cardiomyopathy. Front. Cell Dev. Biol. 2022, 10, 894635. [Google Scholar] [CrossRef]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef]
- Perez-Serra, A.; Toro, R.; Sarquella-Brugada, G.; de Gonzalo-Calvo, D.; Cesar, S.; Carro, E.; Llorente-Cortes, V.; Iglesias, A.; Brugada, J.; Brugada, R.; et al. Genetic basis of dilated cardiomyopathy. Int. J. Cardiol. 2016, 224, 461–472. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; He, J.; Liu, J.; Guo, X.; Chu, H.; Xu, H.; Wang, Y. Comprehensive review on gene mutations contributing to dilated cardiomyopathy. Front. Cardiovasc. Med. 2023, 10, 1296389. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Le Rumeur, E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn. J. Basic. Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef]
- Ben Yaou, R.; Yun, P.; Dabaj, I.; Norato, G.; Donkervoort, S.; Xiong, H.; Nascimento, A.; Maggi, L.; Sarkozy, A.; Monges, S.; et al. International retrospective natural history study of LMNA-related congenital muscular dystrophy. Brain Commun. 2021, 3, fcab075. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Foley, A.R.; Zambon, A.A.; Bonnemann, C.G.; Muntoni, F. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front. Mol. Neurosci. 2020, 13, 123. [Google Scholar] [CrossRef]
- Zambon, A.A.; Muntoni, F. Congenital muscular dystrophies: What is new? Neuromuscul. Disord. 2021, 31, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Chang, S.; Seidman, J.G.; Seidman, C.E. Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Jouk, P.S.; Da Costa, A.; Prieur, F.; Bresson, J.L.; Faivre, L.; Eicher, J.C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med. Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef]
- Schultz, T.I.; Raucci, F.J., Jr.; Salloum, F.N. Cardiovascular Disease in Duchenne Muscular Dystrophy: Overview and Insight Into Novel Therapeutic Targets. JACC Basic. Transl. Sci. 2022, 7, 608–625. [Google Scholar] [CrossRef]
- Ceccato, T.L.; Starbuck, R.B.; Hall, J.K.; Walker, C.J.; Brown, T.E.; Killgore, J.P.; Anseth, K.S.; Leinwand, L.A. Defining the Cardiac Fibroblast Secretome in a Fibrotic Microenvironment. J. Am. Heart Assoc. 2020, 9, e017025. [Google Scholar] [CrossRef]
- Zou, X.; Ouyang, H.; Lin, F.; Zhang, H.; Yang, Y.; Pang, D.; Han, R.; Tang, X. MYBPC3 deficiency in cardiac fibroblasts drives their activation and contributes to fibrosis. Cell Death Dis. 2022, 13, 948. [Google Scholar] [CrossRef]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef]
- Keller, R.S.; Shai, S.Y.; Babbitt, C.J.; Pham, C.G.; Solaro, R.J.; Valencik, M.L.; Loftus, J.C.; Ross, R.S. Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. Am. J. Pathol. 2001, 158, 1079–1090. [Google Scholar] [CrossRef]
- Larson, A.; Codden, C.J.; Huggins, G.S.; Rastegar, H.; Chen, F.Y.; Maron, B.J.; Rowin, E.J.; Maron, M.S.; Chin, M.T. Altered intercellular communication and extracellular matrix signaling as a potential disease mechanism in human hypertrophic cardiomyopathy. Sci. Rep. 2022, 12, 5211. [Google Scholar] [CrossRef]
- Yokota-Nakatsuma, A.; Ohoka, Y.; Takeuchi, H.; Song, S.Y.; Iwata, M. Beta 1-integrin ligation and TLR ligation enhance GM-CSF-induced ALDH1A2 expression in dendritic cells, but differentially regulate their anti-inflammatory properties. Sci. Rep. 2016, 6, 37914. [Google Scholar] [CrossRef]
- Codden, C.J.; Larson, A.; Awata, J.; Perera, G.; Chin, M.T. Single Nucleus RNA-sequencing Reveals Altered Intercellular Communication and Dendritic Cell Activation in Nonobstructive Hypertrophic Cardiomyopathy. Cardiol. Cardiovasc. Med. 2022, 6, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Codden, C.J.; Chin, M.T. Common and Distinctive Intercellular Communication Patterns in Human Obstructive and Nonobstructive Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 946. [Google Scholar] [CrossRef]
- Ewoldt, J.K.; Wang, M.C.; McLellan, M.A.; Cloonan, P.E.; Chopra, A.; Gorham, J.; Li, L.; DeLaughter, D.M.; Gao, X.; Lee, J.H.; et al. Hypertrophic cardiomyopathy-associated mutations drive stromal activation via EGFR-mediated paracrine signaling. Sci. Adv. 2024, 10, eadi6927. [Google Scholar] [CrossRef]
- Heymans, S.; Lakdawala, N.K.; Tschope, C.; Klingel, K. Dilated cardiomyopathy: Causes, mechanisms, and current and future treatment approaches. Lancet 2023, 402, 998–1011. [Google Scholar] [CrossRef]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef]
- Malhotra, R.; Mason, P.K. Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr. Opin. Cardiol. 2009, 24, 203–208. [Google Scholar] [CrossRef]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Merlo, M.; Caiffa, T.; Gobbo, M.; Adamo, L.; Sinagra, G. Reverse remodeling in Dilated Cardiomyopathy: Insights and future perspectives. Int. J. Cardiol. Heart Vasc. 2018, 18, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Rogers, W.J.; Johnstone, D.E.; Yusuf, S.; Weiner, D.H.; Gallagher, P.; Bittner, V.A.; Ahn, S.; Schron, E.; Shumaker, S.A.; Sheffield, L.T. Quality of life among 5025 patients with left ventricular dysfunction randomized between placebo and enalapril: The Studies of Left Ventricular Dysfunction. The SOLVD Investigators. J. Am. Coll. Cardiol. 1994, 23, 393–400. [Google Scholar] [CrossRef]
- Hjalmarson, A.; Goldstein, S.; Fagerberg, B.; Wedel, H.; Waagstein, F.; Kjekshus, J.; Wikstrand, J.; El Allaf, D.; Vitovec, J.; Aldershvile, J.; et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: The Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA 2000, 283, 1295–1302. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef]
- Mistrulli, R.; Ferrera, A.; Salerno, L.; Vannini, F.; Guida, L.; Corradetti, S.; Addeo, L.; Valcher, S.; Di Gioia, G.; Spera, F.R.; et al. Cardiomyopathy and Sudden Cardiac Death: Bridging Clinical Practice with Cutting-Edge Research. Biomedicines 2024, 12, 1602. [Google Scholar] [CrossRef]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An Overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Lin, Z.; Li, Z.; Guo, Z.; Cao, Y.; Li, J.; Liu, P.; Li, Z. Epigenetic Reader Bromodomain Containing Protein 2 Facilitates Pathological Cardiac Hypertrophy via Regulating the Expression of Citrate Cycle Genes. Front. Pharmacol. 2022, 13, 887991. [Google Scholar] [CrossRef]
- Stratton, M.S.; Bagchi, R.A.; Felisbino, M.B.; Hirsch, R.A.; Smith, H.E.; Riching, A.S.; Enyart, B.Y.; Koch, K.A.; Cavasin, M.A.; Alexanian, M.; et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019, 125, 662–677. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wu, R.D.; Lv, Y.G.; Liu, Y.M.; Huang, H.; Xu, J.Q. BRD4 blockage alleviates pathological cardiac hypertrophy through the suppression of fibrosis and inflammation via reducing ROS generation. Biomed. Pharmacother. 2020, 121, 109368. [Google Scholar] [CrossRef]
- Antolic, A.; Wakimoto, H.; Jiao, Z.; Gorham, J.M.; DePalma, S.R.; Lemieux, M.E.; Conner, D.A.; Lee, D.Y.; Qi, J.; Seidman, J.G.; et al. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight 2020, 5, e138687. [Google Scholar] [CrossRef] [PubMed]
- Conejeros, C.; Parra, V.; Sanchez, G.; Pedrozo, Z.; Olmedo, I. Miro1 as a novel regulator of hypertrophy in neonatal rat cardiomyocytes. J. Mol. Cell Cardiol. 2020, 141, 65–69. [Google Scholar] [CrossRef]
- He, Q. Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H210–H216. [Google Scholar] [CrossRef]
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Helio, T.; Kallio, P.; et al. Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 2019, 8, 594. [Google Scholar] [CrossRef]
- Morival, J.L.P.; Widyastuti, H.P.; Nguyen, C.H.H.; Zaragoza, M.V.; Downing, T.L. DNA methylation analysis reveals epimutation hotspots in patients with dilated cardiomyopathy-associated laminopathies. Clin. Epigenetics 2021, 13, 139. [Google Scholar] [CrossRef]
- Chen, S.C.; Kennedy, B.K.; Lampe, P.D. Phosphorylation of connexin43 on S279/282 may contribute to laminopathy-associated conduction defects. Exp. Cell Res. 2013, 319, 888–896. [Google Scholar] [CrossRef]
- Wang, X.; Luo, W.; Chang, J. Deficient Lmna in fibroblasts: An emerging role of non-cardiomyocytes in DCM. J. Cardiovasc. Aging 2022, 2, 38. [Google Scholar] [CrossRef]
- Fairweather, D.; Rose, N.R. Coxsackievirus-induced myocarditis in mice: A model of autoimmune disease for studying immunotoxicity. Methods 2007, 41, 118–122. [Google Scholar] [CrossRef]
- Taylor, L.A.; Carthy, C.M.; Yang, D.; Saad, K.; Wong, D.; Schreiner, G.; Stanton, L.W.; McManus, B.M. Host gene regulation during coxsackievirus B3 infection in mice: Assessment by microarrays. Circ. Res. 2000, 87, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Louzao-Martinez, L.; Vink, A.; Harakalova, M.; Asselbergs, F.W.; Verhaar, M.C.; Cheng, C. Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy. Int. J. Cardiol. 2016, 220, 634–646. [Google Scholar] [CrossRef]
- Prante, C.; Milting, H.; Kassner, A.; Farr, M.; Ambrosius, M.; Schon, S.; Seidler, D.G.; Banayosy, A.E.; Korfer, R.; Kuhn, J.; et al. Transforming growth factor beta1-regulated xylosyltransferase I activity in human cardiac fibroblasts and its impact for myocardial remodeling. J. Biol. Chem. 2007, 282, 26441–26449. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Huang, B.; Yin, C.; Du, Z.; Guo, R.; Nie, Q.; Zhang, Z. Nitro-oleic acid alleviates inflammation and fibrosis by regulating macrophage polarization and fibroblast activation to improve cardiac function in MI mice. Int. Immunopharmacol. 2025, 146, 113710. [Google Scholar] [CrossRef]
- Braumann, S.; Schumacher, W.; Im, N.G.; Nettersheim, F.S.; Mehrkens, D.; Bokredenghel, S.; Hof, A.; Nies, R.J.; Adler, C.; Winkels, H.; et al. Nitro-Oleic Acid (NO(2)-OA) Improves Systolic Function in Dilated Cardiomyopathy by Attenuating Myocardial Fibrosis. Int. J. Mol. Sci. 2021, 22, 9052. [Google Scholar] [CrossRef]
- Perestrelo, A.R.; Silva, A.C.; Oliver-De La Cruz, J.; Martino, F.; Horvath, V.; Caluori, G.; Polansky, O.; Vinarsky, V.; Azzato, G.; de Marco, G.; et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 2021, 128, 24–38. [Google Scholar] [CrossRef]
- Jin, B.; Zhu, J.; Shi, H.M.; Wen, Z.C.; Wu, B.W. YAP activation promotes the transdifferentiation of cardiac fibroblasts to myofibroblasts in matrix remodeling of dilated cardiomyopathy. Braz. J. Med. Biol. Res. 2018, 52, e7914. [Google Scholar] [CrossRef]
- Tsuru, H.; Yoshihara, C.; Suginobe, H.; Matsumoto, M.; Ishii, Y.; Narita, J.; Ishii, R.; Wang, R.; Ueyama, A.; Ueda, K.; et al. Pathogenic Roles of Cardiac Fibroblasts in Pediatric Dilated Cardiomyopathy. J. Am. Heart Assoc. 2023, 12, e029676. [Google Scholar] [CrossRef]
- Lovering, R.M.; Porter, N.C.; Bloch, R.J. The muscular dystrophies: From genes to therapies. Phys. Ther. 2005, 85, 1372–1388. [Google Scholar]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef]
- Cruz Guzman Odel, R.; Chavez Garcia, A.L.; Rodriguez-Cruz, M. Muscular dystrophies at different ages: Metabolic and endocrine alterations. Int. J. Endocrinol. 2012, 2012, 485376. [Google Scholar] [CrossRef]
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Vainzof, M.; Takata, R.I.; Passos-Bueno, M.R.; Pavanello, R.C.; Zatz, M. Is the maintainance of the C-terminus domain of dystrophin enough to ensure a milder Becker muscular dystrophy phenotype? Hum. Mol. Genet. 1993, 2, 39–42. [Google Scholar] [CrossRef]
- Stark, A.E. Determinants of the incidence of Duchenne muscular dystrophy. Ann. Transl. Med. 2015, 3, 287. [Google Scholar] [CrossRef]
- Acsadi, G.; Crawford, T.O.; Muller-Felber, W.; Shieh, P.B.; Richardson, R.; Natarajan, N.; Castro, D.; Ramirez-Schrempp, D.; Gambino, G.; Sun, P.; et al. Safety and efficacy of nusinersen in spinal muscular atrophy: The EMBRACE study. Muscle Nerve 2021, 63, 668–677. [Google Scholar] [CrossRef]
- Mercuri, E.; Pane, M.; Cicala, G.; Brogna, C.; Ciafaloni, E. Detecting early signs in Duchenne muscular dystrophy: Comprehensive review and diagnostic implications. Front. Pediatr. 2023, 11, 1276144. [Google Scholar] [CrossRef]
- James, J.; Kinnett, K.; Wang, Y.; Ittenbach, R.F.; Benson, D.W.; Cripe, L. Electrocardiographic abnormalities in very young Duchenne muscular dystrophy patients precede the onset of cardiac dysfunction. Neuromuscul. Disord. 2011, 21, 462–467. [Google Scholar] [CrossRef]
- Ke, Q.; Zhao, Z.Y.; Mendell, J.R.; Baker, M.; Wiley, V.; Kwon, J.M.; Alfano, L.N.; Connolly, A.M.; Jay, C.; Polari, H.; et al. Progress in treatment and newborn screening for Duchenne muscular dystrophy and spinal muscular atrophy. World J. Pediatr. 2019, 15, 219–225. [Google Scholar] [CrossRef]
- Saad, F.A.; Siciliano, G.; Angelini, C. Advances in Dystrophinopathy Diagnosis and Therapy. Biomolecules 2023, 13, 1319. [Google Scholar] [CrossRef]
- Santos, M.A.; Costa Fde, A.; Travessa, A.F.; Bombig, M.T.; Fonseca, F.H.; Luna Filho, B.; Mussi, A.; Souza, D.; Oliveira, A.; Povoa, R. [Duchenne muscular dystrophy: Electrocardiographic analysis of 131 patients]. Arq. Bras. Cardiol. 2010, 94, 620–624. [Google Scholar] [CrossRef]
- Sahani, R.; Wallace, C.H.; Jones, B.K.; Blemker, S.S. Diaphragm muscle fibrosis involves changes in collagen organization with mechanical implications in Duchenne muscular dystrophy. J. Appl. Physiol. 2022, 132, 653–672. [Google Scholar] [CrossRef]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef]
- Zanotti, S.; Gibertini, S.; Bragato, C.; Mantegazza, R.; Morandi, L.; Mora, M. Fibroblasts from the muscles of Duchenne muscular dystrophy patients are resistant to cell detachment apoptosis. Exp. Cell Res. 2011, 317, 2536–2547. [Google Scholar] [CrossRef]
- Soussi, S.; Savchenko, L.; Rovina, D.; Iacovoni, J.S.; Gottinger, A.; Vialettes, M.; Pioner, J.M.; Farini, A.; Mallia, S.; Rabino, M.; et al. IPSC derived cardiac fibroblasts of DMD patients show compromised actin microfilaments, metabolic shift and pro-fibrotic phenotype. Biol. Direct 2023, 18, 41. [Google Scholar] [CrossRef]
- Zanotti, S.; Gibertini, S.; Blasevich, F.; Bragato, C.; Ruggieri, A.; Saredi, S.; Fabbri, M.; Bernasconi, P.; Maggi, L.; Mantegazza, R.; et al. Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol. 2018, 74, 77–100. [Google Scholar] [CrossRef]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef]
- Ertl, G.; Frantz, S. Healing after myocardial infarction. Cardiovasc. Res. 2005, 66, 22–32. [Google Scholar] [CrossRef]
- Webber, M.; Jackson, S.P.; Moon, J.C.; Captur, G. Myocardial Fibrosis in Heart Failure: Anti-Fibrotic Therapies and the Role of Cardiovascular Magnetic Resonance in Drug Trials. Cardiol. Ther. 2020, 9, 363–376. [Google Scholar] [CrossRef]
- Neefs, J.; van den Berg, N.W.; Limpens, J.; Berger, W.R.; Boekholdt, S.M.; Sanders, P.; de Groot, J.R. Aldosterone Pathway Blockade to Prevent Atrial Fibrillation: A Systematic Review and Meta-Analysis. Int. J. Cardiol. 2017, 231, 155–161. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, C.; Song, Q.; Li, Y.; Da, X.; Yu, Y.; Li, H.; Clark, I.M.; Chen, Q.; Wang, Q.K. ADAMTS16 activates latent TGF-beta, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc. Res. 2020, 116, 956–969. [Google Scholar] [CrossRef]
- Humeres, C.; Shinde, A.V.; Hanna, A.; Alex, L.; Hernandez, S.C.; Li, R.; Chen, B.; Conway, S.J.; Frangogiannis, N.G. Smad7 effects on TGF-beta and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 2022, 132, e146926. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.Y.; Liu, Z.Q.; Jiang, D.; Wu, S.Y.; Guo, Y.Q.; Tao, H.M.; Sun, M.; You, L.N.; Qin, S.; et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-beta1/Smads and activating P53 signaling pathways. Cell Death Dis. 2020, 11, 44. [Google Scholar] [CrossRef]
- Korf-Klingebiel, M.; Reboll, M.R.; Klede, S.; Brod, T.; Pich, A.; Polten, F.; Napp, L.C.; Bauersachs, J.; Ganser, A.; Brinkmann, E.; et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat. Med. 2015, 21, 140–149. [Google Scholar] [CrossRef]
- Shyu, K.G. The Role of Endoglin in Myocardial Fibrosis. Acta Cardiol. Sin. 2017, 33, 461–467. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Hinderer, S.; Brauchle, E.; Schenke-Layland, K. Generation and Assessment of Functional Biomaterial Scaffolds for Applications in Cardiovascular Tissue Engineering and Regenerative Medicine. Adv. Healthc. Mater. 2015, 4, 2326–2341. [Google Scholar] [CrossRef]
- Christman, K.L.; Lee, R.J. Biomaterials for the treatment of myocardial infarction. J. Am. Coll. Cardiol. 2006, 48, 907–913. [Google Scholar] [CrossRef]
- Zhong, Y.; Yang, Y.; Xu, Y.; Qian, B.; Huang, S.; Long, Q.; Qi, Z.; He, X.; Zhang, Y.; Li, L.; et al. Design of a Zn-based nanozyme injectable multifunctional hydrogel with ROS scavenging activity for myocardial infarction therapy. Acta Biomater. 2024, 177, 62–76. [Google Scholar] [CrossRef]
- Hinderer, S.; Schesny, M.; Bayrak, A.; Ibold, B.; Hampel, M.; Walles, T.; Stock, U.A.; Seifert, M.; Schenke-Layland, K. Engineering of fibrillar decorin matrices for a tissue-engineered trachea. Biomaterials 2012, 33, 5259–5266. [Google Scholar] [CrossRef]
- Hinderer, S.; Seifert, J.; Votteler, M.; Shen, N.; Rheinlaender, J.; Schaffer, T.E.; Schenke-Layland, K. Engineering of a bio-functionalized hybrid off-the-shelf heart valve. Biomaterials 2014, 35, 2130–2139. [Google Scholar] [CrossRef]
- Monaghan, M.; Browne, S.; Schenke-Layland, K.; Pandit, A. A collagen-based scaffold delivering exogenous microrna-29B to modulate extracellular matrix remodeling. Mol. Ther. 2014, 22, 786–796. [Google Scholar] [CrossRef]
- Miyagawa, S.; Domae, K.; Yoshikawa, Y.; Fukushima, S.; Nakamura, T.; Saito, A.; Sakata, Y.; Hamada, S.; Toda, K.; Pak, K.; et al. Phase I Clinical Trial of Autologous Stem Cell-Sheet Transplantation Therapy for Treating Cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e003918. [Google Scholar] [CrossRef]
- Cilvik, S.N.; Wang, J.I.; Lavine, K.J.; Uchida, K.; Castro, A.; Gierasch, C.M.; Weinheimer, C.J.; House, S.L.; Kovacs, A.; Nichols, C.G.; et al. Fibroblast growth factor receptor 1 signaling in adult cardiomyocytes increases contractility and results in a hypertrophic cardiomyopathy. PLoS ONE 2013, 8, e82979. [Google Scholar] [CrossRef]
- Gao, Z.; Yan, L.; Meng, J.; Lu, Z.; Ge, K.; Jiang, Z.; Feng, T.; Wang, H.; Liu, C.; Tang, J.; et al. Targeting cardiac fibrosis with chimeric antigen receptor macrophages. Cell Discov. 2024, 10, 86. [Google Scholar] [CrossRef]
- Varzideh, F.; Kansakar, U.; Donkor, K.; Wilson, S.; Jankauskas, S.S.; Mone, P.; Wang, X.; Lombardi, A.; Santulli, G. Cardiac Remodeling After Myocardial Infarction: Functional Contribution of microRNAs to Inflammation and Fibrosis. Front. Cardiovasc. Med. 2022, 9, 863238. [Google Scholar] [CrossRef]
- O’Reilly, S. MicroRNAs in fibrosis: Opportunities and challenges. Arthritis Res. Ther. 2016, 18, 11. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Deng, Y.; Li, H. MicroRNA-223 Regulates Cardiac Fibrosis After Myocardial Infarction by Targeting RASA1. Cell Physiol. Biochem. 2018, 46, 1439–1454. [Google Scholar] [CrossRef]
- Liu, Y.; Song, J.W.; Lin, J.Y.; Miao, R.; Zhong, J.C. Roles of MicroRNA-122 in Cardiovascular Fibrosis and Related Diseases. Cardiovasc. Toxicol. 2020, 20, 463–473. [Google Scholar] [CrossRef]
- Shen, J.; Xing, W.; Gong, F.; Wang, W.; Yan, Y.; Zhang, Y.; Xie, C.; Fu, S. MiR-150-5p retards the progression of myocardial fibrosis by targeting EGR1. Cell Cycle 2019, 18, 1335–1348. [Google Scholar] [CrossRef]
- Chiasson, V.; Takano, A.P.C.; Guleria, R.S.; Gupta, S. Deficiency of MicroRNA miR-1954 Promotes Cardiac Remodeling and Fibrosis. J. Am. Heart Assoc. 2019, 8, e012880. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, T.; Zhang, S. MicroRNA-489 suppresses isoproterenol-induced cardiac fibrosis by downregulating histone deacetylase 2. Exp. Ther. Med. 2020, 19, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, L.; Wu, X.; Yi, H. RSPO4-CRISPR alleviates liver injury and restores gut microbiota in a rat model of liver fibrosis. Commun. Biol. 2021, 4, 230. [Google Scholar] [CrossRef]
- Nishiga, M.; Liu, C.; Qi, L.S.; Wu, J.C. The use of new CRISPR tools in cardiovascular research and medicine. Nat. Rev. Cardiol. 2022, 19, 505–521. [Google Scholar] [CrossRef]
- Park, H.; Kim, D.; Cho, B.; Byun, J.; Kim, Y.S.; Ahn, Y.; Hur, J.; Oh, Y.K.; Kim, J. In vivo therapeutic genome editing via CRISPR/Cas9 magnetoplexes for myocardial infarction. Biomaterials 2022, 281, 121327. [Google Scholar] [CrossRef]
- Jiang, L.; Liang, J.; Huang, W.; Ma, J.; Park, K.H.; Wu, Z.; Chen, P.; Zhu, H.; Ma, J.J.; Cai, W.; et al. CRISPR activation of endogenous genes reprograms fibroblasts into cardiovascular progenitor cells for myocardial infarction therapy. Mol. Ther. 2022, 30, 54–74. [Google Scholar] [CrossRef]
- Cho, H.M.; Lee, K.H.; Shen, Y.M.; Shin, T.J.; Ryu, P.D.; Choi, M.C.; Kang, K.S.; Cho, J.Y. Transplantation of hMSCs Genome Edited with LEF1 Improves Cardio-Protective Effects in Myocardial Infarction. Mol. Ther. Nucleic Acids 2020, 19, 1186–1197. [Google Scholar] [CrossRef]
- Meng, X.; Zheng, M.; Yu, M.; Bai, W.; Zuo, L.; Bu, X.; Liu, Y.; Xia, L.; Hu, J.; Liu, L.; et al. Transplantation of CRISPRa system engineered IL10-overexpressing bone marrow-derived mesenchymal stem cells for the treatment of myocardial infarction in diabetic mice. J. Biol. Eng. 2019, 13, 49. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, J.; Wu, M.; Kang, L.; Xu, B. The effector cells and cellular mediators of immune system involved in cardiac inflammation and fibrosis after myocardial infarction. J. Cell Physiol. 2020, 235, 8996–9004. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Abdalla, A.M.E.; Miao, Y.; Ahmed, A.I.M.; Meng, N.; Ouyang, C. CAR-T cell therapeutic avenue for fighting cardiac fibrosis: Roadblocks and perspectives. Cell Biochem. Funct. 2024, 42, e3955. [Google Scholar] [CrossRef]
- Rurik, J.G.; Tombacz, I.; Yadegari, A.; Mendez Fernandez, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Kablak-Ziembicka, A.; Badacz, R.; Okarski, M.; Wawak, M.; Przewlocki, T.; Podolec, J. Cardiac microRNAs: Diagnostic and therapeutic potential. Arch. Med. Sci. 2023, 19, 1360–1381. [Google Scholar] [CrossRef]
- Singh, B.N.; Koyano-Nakagawa, N.; Gong, W.; Moskowitz, I.P.; Weaver, C.V.; Braunlin, E.; Das, S.; van Berlo, J.H.; Garry, M.G.; Garry, D.J. A conserved HH-Gli1-Mycn network regulates heart regeneration from newt to human. Nat. Commun. 2018, 9, 4237. [Google Scholar] [CrossRef]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef]
- Magadum, A. Modified mRNA Therapeutics for Heart Diseases. Int. J. Mol. Sci. 2022, 23, 15514. [Google Scholar] [CrossRef]
- Kaur, K.; Hadas, Y.; Kurian, A.A.; Zak, M.M.; Yoo, J.; Mahmood, A.; Girard, H.; Komargodski, R.; Io, T.; Santini, M.P.; et al. Direct reprogramming induces vascular regeneration post muscle ischemic injury. Mol. Ther. 2021, 29, 3042–3058. [Google Scholar] [CrossRef]
- Labanieh, L.; Mackall, C.L. CAR immune cells: Design principles, resistance and the next generation. Nature 2023, 614, 635–648. [Google Scholar] [CrossRef]
- Zhang, X.; Song, W.; Qin, C.; Song, Y.; Liu, F.; Hu, F.; Lan, X. Uterine Uptake of 68Ga-FAPI-04 in Uterine Pathology and Physiology. Clin. Nucl. Med. 2022, 47, 7–13. [Google Scholar] [CrossRef]
- Koenig, A.L.; Shchukina, I.; Amrute, J.; Andhey, P.S.; Zaitsev, K.; Lai, L.; Bajpai, G.; Bredemeyer, A.; Smith, G.; Jones, C.; et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 2022, 1, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Farrukh, H.; Chittepu, V.; Xu, H.; Pan, C.X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers/Factors | Embryonic Fibroblast | Neonatal Fibroblast | Adult Fibroblast | References |
|---|---|---|---|---|
| Tcf21 | - | + | + | [22,25] |
| Tbx20 | + | + | + | [26,27] |
| FSP1 | - | + | + | [25,28] |
| Prolyl-4Hydroxylase | - | - | + | [29,30] |
| Vimentin | - | + | + | [31,32] |
| αSMA | - | - | - | [32,33] |
| PDGFRα | - | + | + | [2,31,34] |
| MEFSK4 | - | - | + | [21] |
| DDR2 | - | + | + | [25,35] |
| CD90 | - | + | + | [21,36] |
| Sca1 | - | - | + | [26,37] |
| Periostin | - | + | - | [25,32,38] |
| Fibronectin | - | + | + | [32,39] |
| Collagen type I | - | + | + | [25,31] |
| Collagen type III | - | + | + | [25] |
| Disease | Gene Mutation | FB Activation | Outcomes | References |
|---|---|---|---|---|
| Hypertrophic Cardiomyopathy | MYH7, MYBP3 | Activated | Contractile dysfunction due to the stiffening and thickening of the left ventricular, myocyte disarray and hypertrophy, heart failure or sudden cardiac death. | [122,123,124,125] |
| Dilated Cardiomyopathy | TTN, LMN, EEF1A2, SYNE1/SYNE2, PRDM16 | Activated | Contractile dysfunction due to ventricular dilatation, heart failure or sudden cardiac death. | [123,126,127,128] |
| Duchene muscular dystrophy (DMD) | Dystrophin | Activated | Intracellular Ca2+ ion increases, myofiber atrophy/fibrosis, loss of muscle function, cardiac problems, respiratory dysfunctions, death between 20 and 40 years of age. | [129] |
| Becker muscular dystrophy (BMD) | Dystrophin | Activated | Milder than DMD and progresses slowly, Muscle weakness as in DMD. | [130] |
| Congenital Muscular Dystrophy (CMD) | Merosin, LMNA, DAG1 | Activated | Joint contractures, cognitive and speech problems, seizures. | [131,132,133] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shameem, M.; Olson, S.L.; Marron Fernandez de Velasco, E.; Kumar, A.; Singh, B.N. Cardiac Fibroblasts: Helping or Hurting. Genes 2025, 16, 381. https://doi.org/10.3390/genes16040381
Shameem M, Olson SL, Marron Fernandez de Velasco E, Kumar A, Singh BN. Cardiac Fibroblasts: Helping or Hurting. Genes. 2025; 16(4):381. https://doi.org/10.3390/genes16040381
Chicago/Turabian StyleShameem, Mohammad, Shelby L. Olson, Ezequiel Marron Fernandez de Velasco, Akhilesh Kumar, and Bhairab N. Singh. 2025. "Cardiac Fibroblasts: Helping or Hurting" Genes 16, no. 4: 381. https://doi.org/10.3390/genes16040381
APA StyleShameem, M., Olson, S. L., Marron Fernandez de Velasco, E., Kumar, A., & Singh, B. N. (2025). Cardiac Fibroblasts: Helping or Hurting. Genes, 16(4), 381. https://doi.org/10.3390/genes16040381