
Novel Filaggrin Variants Are Associated with Ichthyosis Vulgaris in Mexicans

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Material and Methods
2.1. Participants
2.2. STS Activity
2.3. Automated Sequencing
2.4. Ethical Aspects
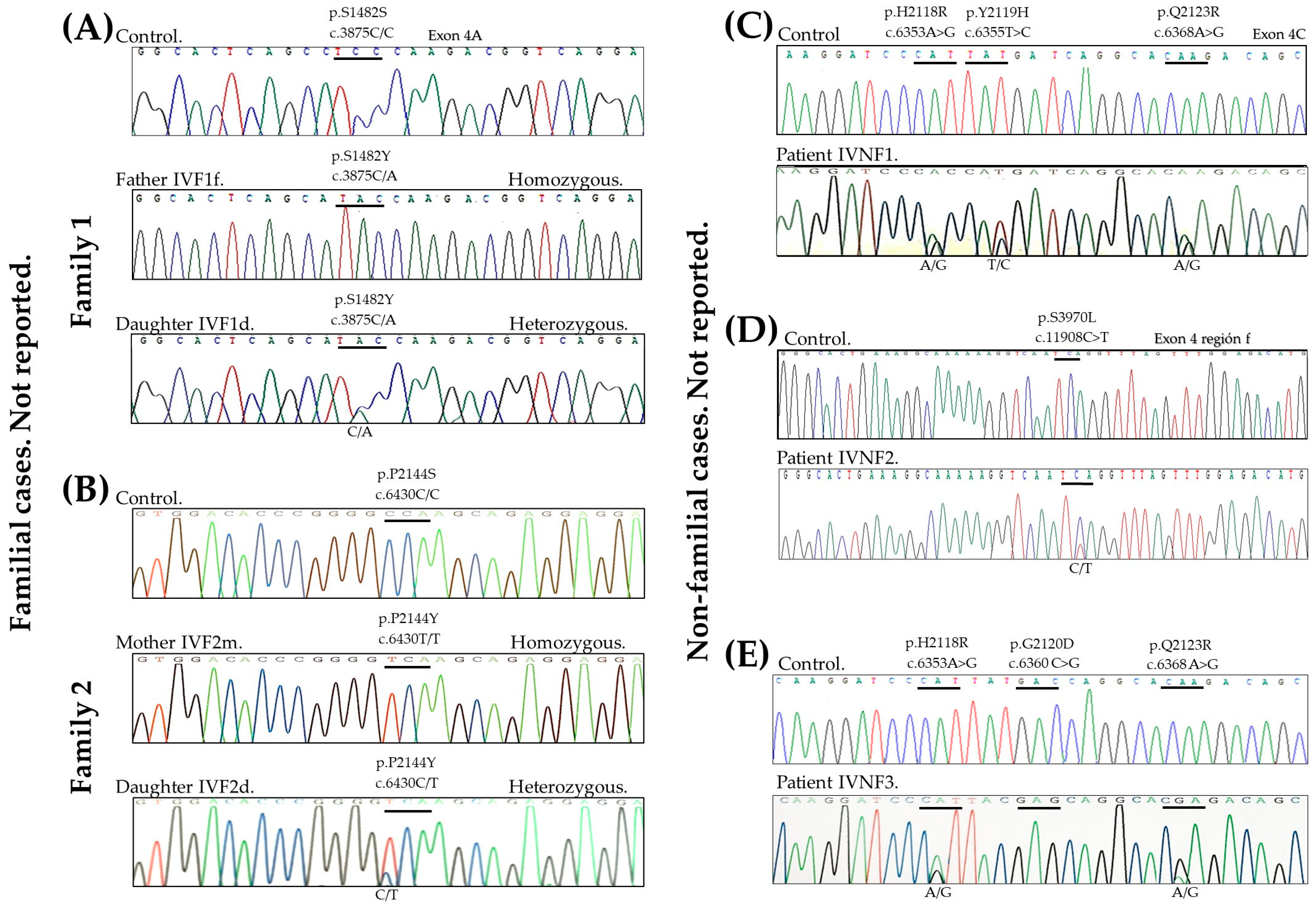
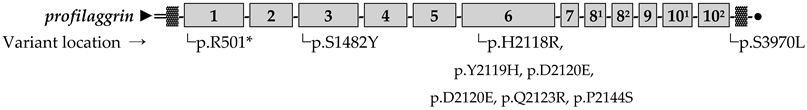
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vega Almendra, N.; Aranibar Duran, L. Hereditary ichthyosis: A diagnotic and therapeutic challenge. Rev. Chil. Pediatr. 2016, 87, 213–223. [Google Scholar]
- Gutiérrez-Cerrajero, C.; Sprecher, E.; Paller, A.S.; Akiyama, M.; Mazereeuw-Hautier, J.; Hernández-Martín, A.; González-Sarmiento, R. Ichthyosis. Nat. Rev. Dis. Primers 2023, 19, 2. [Google Scholar]
- Takeichi, T.; Akiyama, M. Inherited ichthyosis: Non-syndromic forms. J. Dermatol. 2016, 43, 242–251. [Google Scholar] [PubMed]
- Brown, S.J.; Relton, C.L.; Liao, H.; Zhao, Y.; Sandilands, A.; Wilson, I.J.; Burn, J.; Reynolds, N.J.; McLean, W.H.; Cordell, H.J. Filaggrin null mutations and childhood atopic eczema: A population-based case-control study. J. Allergy Clin. Immunol. 2008, 121, 940–946. [Google Scholar] [PubMed]
- Smith, F.J.; Irvine, A.D.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342. [Google Scholar]
- Wells, R.S.; Kerr, C.B. Clinical features of autosomal dominant and sex-linked ichthyosis in an English population. Br. Med. J. 1966, 1, 947–950. [Google Scholar]
- Sybert, V.P.; Dale, B.A.; Holbrook, K.A. Ichthyosis vulgaris: Identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules. J. Investig. Dermatol. 1985, 84, 191–194. [Google Scholar]
- Thyssen, J.P.; Godoy-Gijon, E.; Elias, P.M. Ichthyosis vulgaris: The filaggrin mutation disease. Br. J. Dermatol. 2013, 168, 1155–1166. [Google Scholar] [CrossRef]
- Oji, V.; Traupe, H. Ichthyoses: Differential diagnosis and molecular genetics. Eur. J. Dermatol. 2006, 16, 349–359. [Google Scholar]
- Oji, V. Clinical presentation and etiology of ichthyoses. Overview of the new nomenclature and classification. Hautarzt 2010, 61, 891–902; quiz 903–904. [Google Scholar]
- Greisenegger, E.K.; Llufriu, S.; Chamorro, A.; Cervera, A.; Jimenez-Escrig, A.; Rappersberger, K.; Marik, W.; Greisenegger, S.; Stögmann, E.; Kopp, T.; et al. A NOTCH3 homozygous nonsense mutation in familial Sneddon syndrome with pediatric stroke. J. Neurol. 2021, 268, 810–816. [Google Scholar] [PubMed]
- Sandilands, A.; O’Regan, G.M.; Liao, H.; Zhao, Y.; Terron-Kwiatkowski, A.; Watson, R.M.; Cassidy, A.J.; Goudie, D.R.; Smith, F.J.; McLean, W.H.; et al. Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J. Investig. Dermatol. 2006, 126, 1770–1775. [Google Scholar] [PubMed]
- Armengot-Carbo, M.; Hernández-Martín, Á.; Torrelo, A. The role of filaggrin in the skin barrier and disease development. Actas Dermosifiliogr. 2015, 106, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Jeong, M.S.; Li, K.; Park, M.K.; Lee, M.K.; Yoon, Y.; Cho, D.Y.; Seo, S.J. Genetic Polymorphism of FLG in Korean Ichthyosis Vulgaris Patients. Ann. Dermatol. 2011, 23, 170–176. [Google Scholar] [CrossRef]
- Park, K.Y.; Li, K.; Seok, J.; Seo, S.J. An Analysis of the Filaggrin Gene Polymorphism in Korean Atopic Dermatitis Patients. J. Korean Med. Sci. 2016, 31, 1136–1142. [Google Scholar] [CrossRef]
- Wong, X.F.C.C.; Denil, S.L.I.J.; Foo, J.N.; Chen, H.; Tay, A.S.L.; Haines, R.L.; Tang, M.B.Y.; McLean, W.H.I.; Sandilands, A.; Smith, F.J.D.; et al. Array-based sequencing of filaggrin gene for comprehensive detection of disease-associated variants. J. Allergy Clin. Immunol. 2018, 141, 814–816. [Google Scholar] [CrossRef]
- Kiritsi, D.; Valari, M.; Fortugno, P.; Hausser, I.; Lykopoulou, L.; Zambruno, G.; Fischer, J.; Bruckner-Tuderman, L.; Jakob, T.; Has, C. Whole-exome sequencing in patients with ichthyosis reveals modifiers associated with increased IgE levels and allergic sensitizations. J. Allergy Clin. Immunol. 2015, 135, 280–283. [Google Scholar] [CrossRef]
- Chang, Y.C.; Wu, W.M.; Chen, C.H.; Hu, C.F.; Hsu, L.A. Association between P478S polymorphism of the filaggrin gene and risk of psoriasis in a Chinese population in Taiwan. Arch. Dermatol. Res. 2008, 300, 133–137. [Google Scholar] [CrossRef]
- Hassani, B.; Isaian, A.; Shariat, M.; Mollanoori, H.; Sotoudeh, S.; Babaei, V.; Ziaali, A.; Teimourian, S. Filaggrin gene polymorphisms in Iranian ichthyosis vulgaris and atopic dermatitis patients. Int. J. Dermatol. 2018, 57, 1485–1491. [Google Scholar] [CrossRef]
- Harding, C.R.; Aho, S.; Bosko, C.A. Filaggrin—Revisited. Int. J. Cosmet. Sci. 2013, 35, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Hoober, J.K.; Eggink, L.L. The Discovery and Function of Filaggrin. Int. J. Mol. Sci. 2022, 23, 1455. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liang, C.; Li, P.; Xiao, H. Revisiting X-linked congenital ichthyosis. Int. J. Dermatol. 2025, 64, 51–61. [Google Scholar] [PubMed]
- Cuevas-Covarrubias, S.A.; Jiménez-Vaca, A.L.; González-Huerta, L.M.; Rivera-Vega, M.d.R.; Kofman-Alfaro, S.H.; Valdes-Flores, M.; Maya-Nunez, G. Somatic and germinal mosaicism for the steroid sulfatase gene deletion in a steroid sulfatase deficiency carrier. J. Investig. Dermatol. 2002, 119, 972–975. [Google Scholar] [PubMed]
- Alakloby, O.M.; Almuqarrab, F.; Zschocke, J.; Schmuth, M.; Abdulkareem, A.; Alnutaifi, K.; Borgio, F.; Gruber, R.; Hennies, H.C. Filaggrin gene variants among Saudi patients with ichthyosis vulgaris. BMC Med. Genom. 2023, 16, 256. [Google Scholar] [CrossRef]
- NOM-253-SSA1-2012; Para la Disposición de Sangre Humana y sus Componentes con Fines Terapéuticos. Comité Consultivo Nacional de Normalización de Regulacion y Fomento Sanitario (SSA1): México City, Mexico, 2012.
- Margolis, D.J.; Mitra, N.; Wubbenhorst, B.; D’Andrea, K.; Kraya, A.A.; Hoffstad, O.; Shah, S.; Nathanson, K.L. Association of Filaggrin Loss-of-Function Variants with Race in Children with Atopic Dermatitis. JAMA Dermatol. 2019, 155, 1269–1276. [Google Scholar]


{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Family Cases | Participant Record | Protein | Nucleotide | Control | Change Allele Found in Patient | CV Variation ID | References |
| Family 1 | IVF1f father | p.S1482Y | c.4445C>A | TCC | TAC, TAC | 1277512 | NR |
| IVF1d daughter | TCC | TCC, TAC | |||||
| Family 2 | IVF2m mother | p.P2144S | c.6430C>T | CCA | TCA, TCA | 1206192 | NR |
| IVF2d daughter | CCA | CCA, TCA | |||||
| No | IVNF1 | p.H2118R p.Y2119H p.Q2123R | c.6353A>G c.6355T>C c.6368A>G | CAT TAT CAA | CAT, CGC TAT, CAT CAA, CGA | NR 1221916 NR | NR Margolis, 2019 [26] NR |
| No | IVNF2 | p.S3970L | c.11908C>T | TCA | TCA, TTA | 1231239 | NR |
| No | IVNF3 | p.H2118R p.D2120E p.Q2123R | c.6353A>G c.6360C>G c.6368A>G | CAT GAC CAA | CAT, CGT GAC, GAG CAA, CGA | NR NR NR | NR NR NR |
| None | IVNF4 to IVNF18 | p.R501* | c.1501C>T | CGA | TGA, TGA | 16319 | Smith FJ, 2006 [5] |
 FLG repeats. ▓ Imperfect FLG repeats. ● C-terminal domain.
FLG repeats. ▓ Imperfect FLG repeats. ● C-terminal domain.Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Huerta, L.M.; Zúñiga-Rodríguez, F.G.; Valerio-Gómez, V.I.; Velasco-Medina, A.A.; del Refugio Rivera-Vega, M.; Hernández-Zamora, E.; Toral-López, J. Novel Filaggrin Variants Are Associated with Ichthyosis Vulgaris in Mexicans. Genes 2025, 16, 380. https://doi.org/10.3390/genes16040380
González-Huerta LM, Zúñiga-Rodríguez FG, Valerio-Gómez VI, Velasco-Medina AA, del Refugio Rivera-Vega M, Hernández-Zamora E, Toral-López J. Novel Filaggrin Variants Are Associated with Ichthyosis Vulgaris in Mexicans. Genes. 2025; 16(4):380. https://doi.org/10.3390/genes16040380
Chicago/Turabian StyleGonzález-Huerta, Luz María, Francisco Gabino Zúñiga-Rodríguez, Valeria Isabel Valerio-Gómez, Andrea Aida Velasco-Medina, María del Refugio Rivera-Vega, Edgar Hernández-Zamora, and Jaime Toral-López. 2025. "Novel Filaggrin Variants Are Associated with Ichthyosis Vulgaris in Mexicans" Genes 16, no. 4: 380. https://doi.org/10.3390/genes16040380
APA StyleGonzález-Huerta, L. M., Zúñiga-Rodríguez, F. G., Valerio-Gómez, V. I., Velasco-Medina, A. A., del Refugio Rivera-Vega, M., Hernández-Zamora, E., & Toral-López, J. (2025). Novel Filaggrin Variants Are Associated with Ichthyosis Vulgaris in Mexicans. Genes, 16(4), 380. https://doi.org/10.3390/genes16040380