
Genome-Wide Runs of Homozygosity Reveal Inbreeding Levels and Trait-Associated Candidate Genes in Diverse Sheep Breeds

Abstract
1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequence Data
2.2. Read Alignment and SNP Calling
2.3. Population Genetic Analysis
2.4. Identification of ROH
2.5. Calculation of Inbreeding Coefficient
2.6. Detection of ROH Islands and Candidate Genes for Various Traits
3. Results
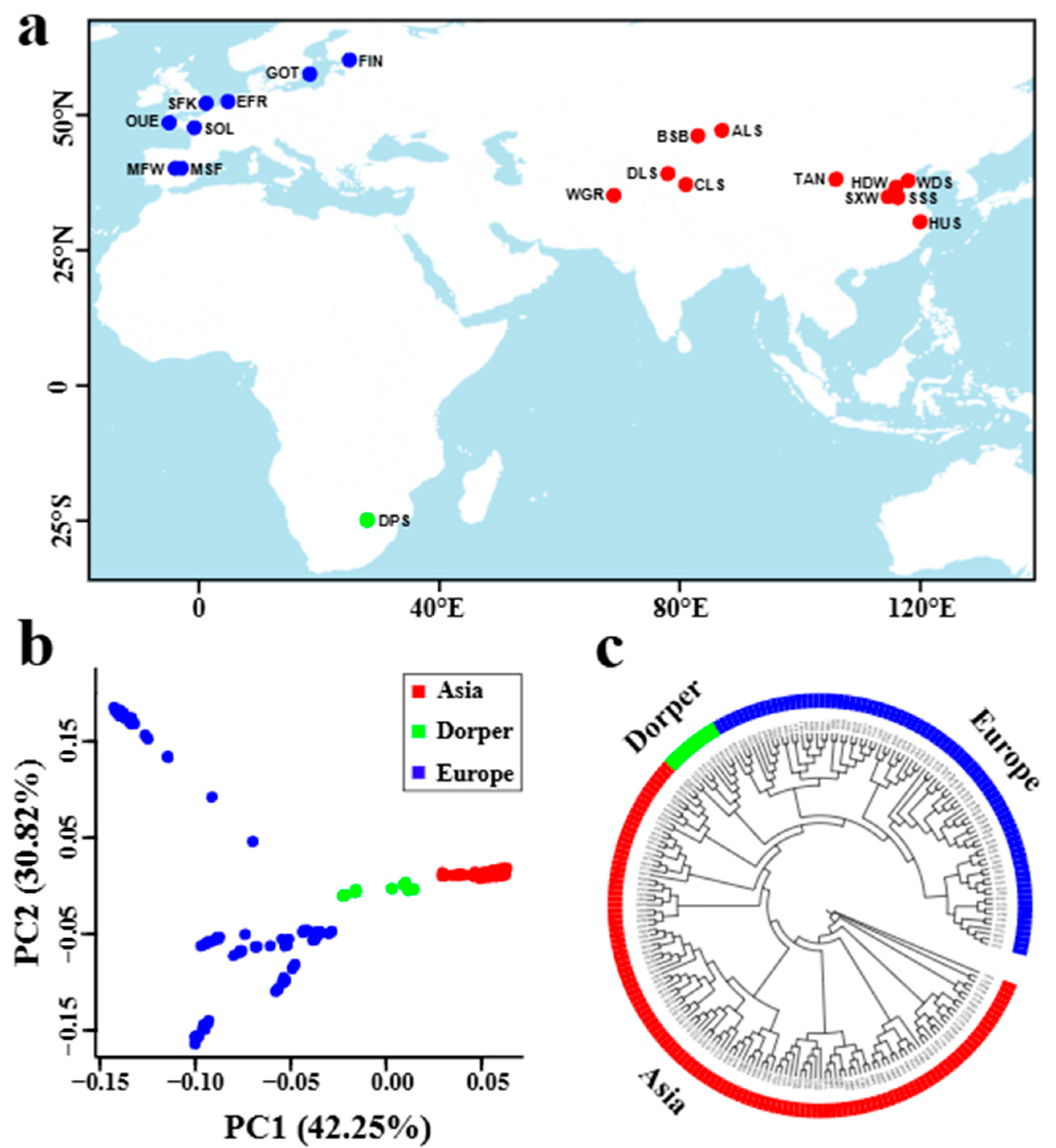
3.1. Population Structure
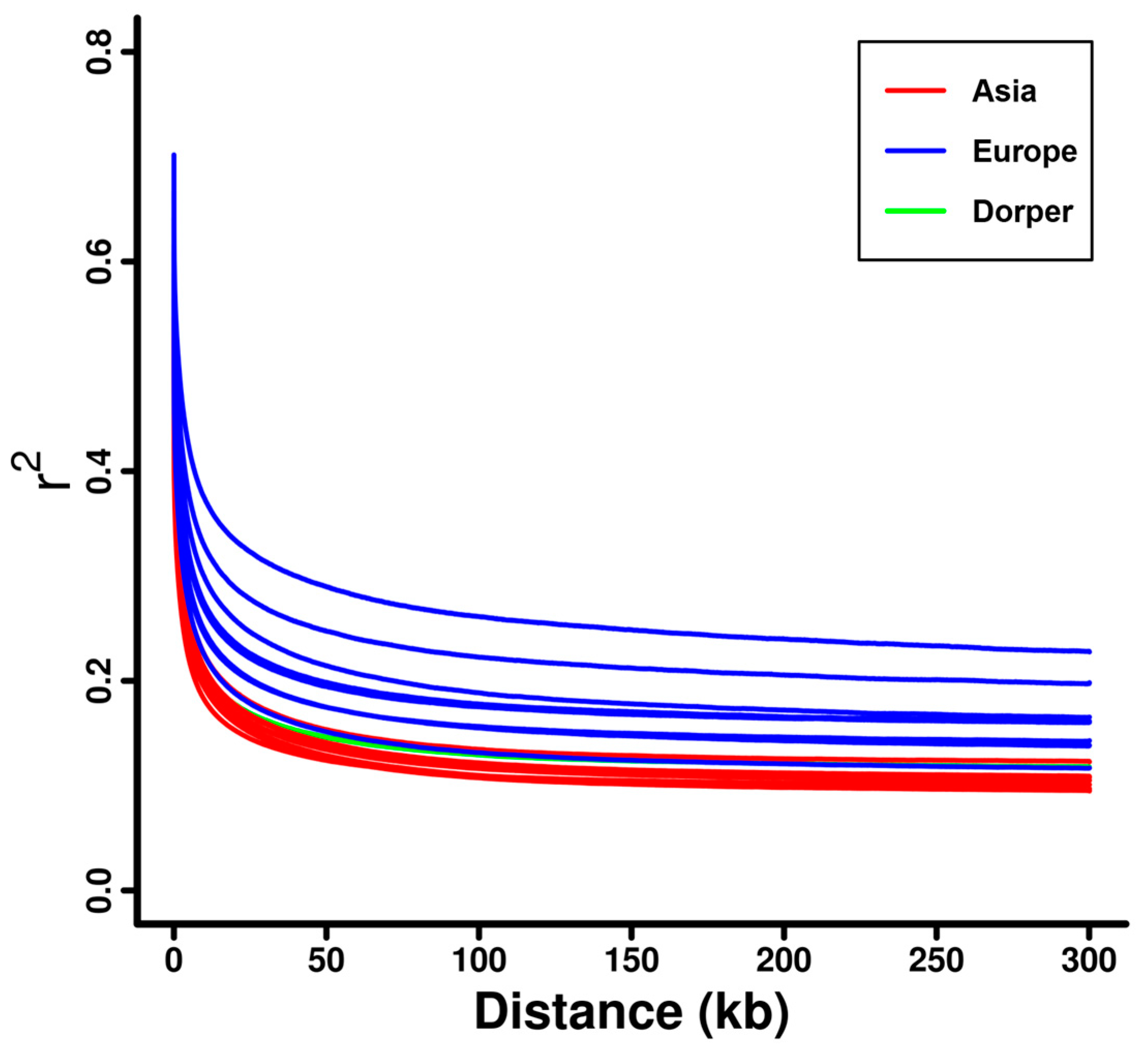
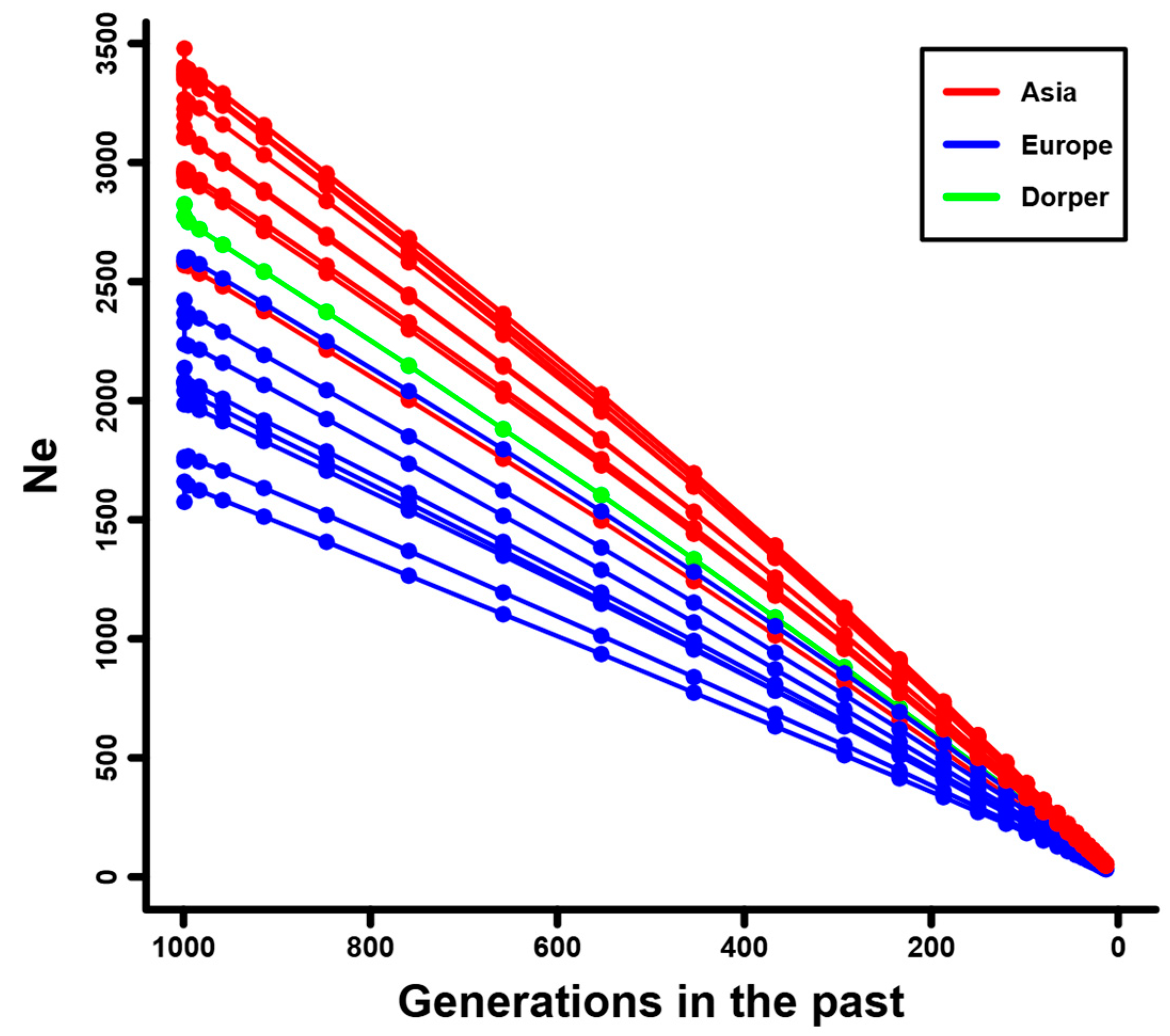
3.2. Linkage Disequilibrium and Effective Population Size
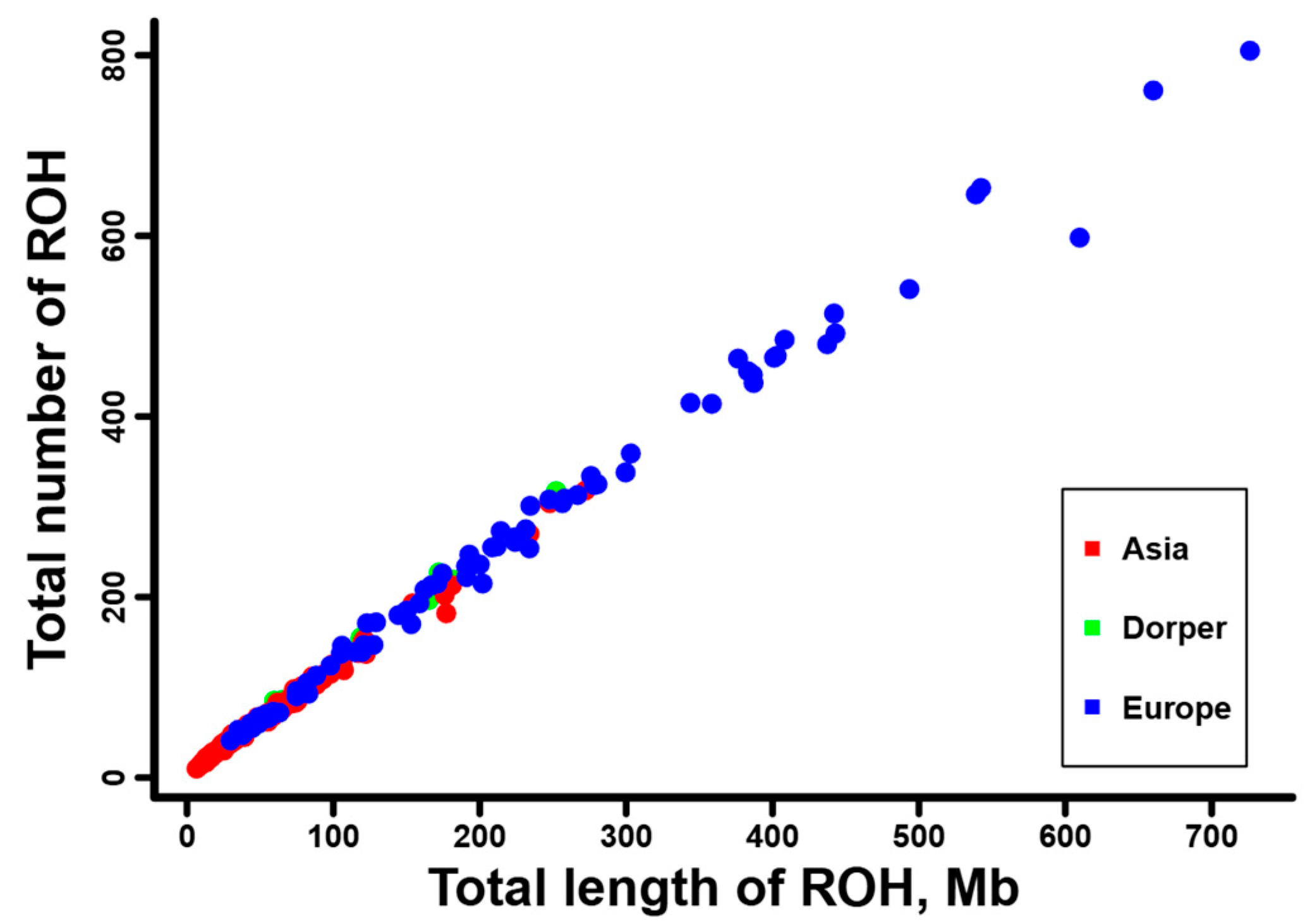
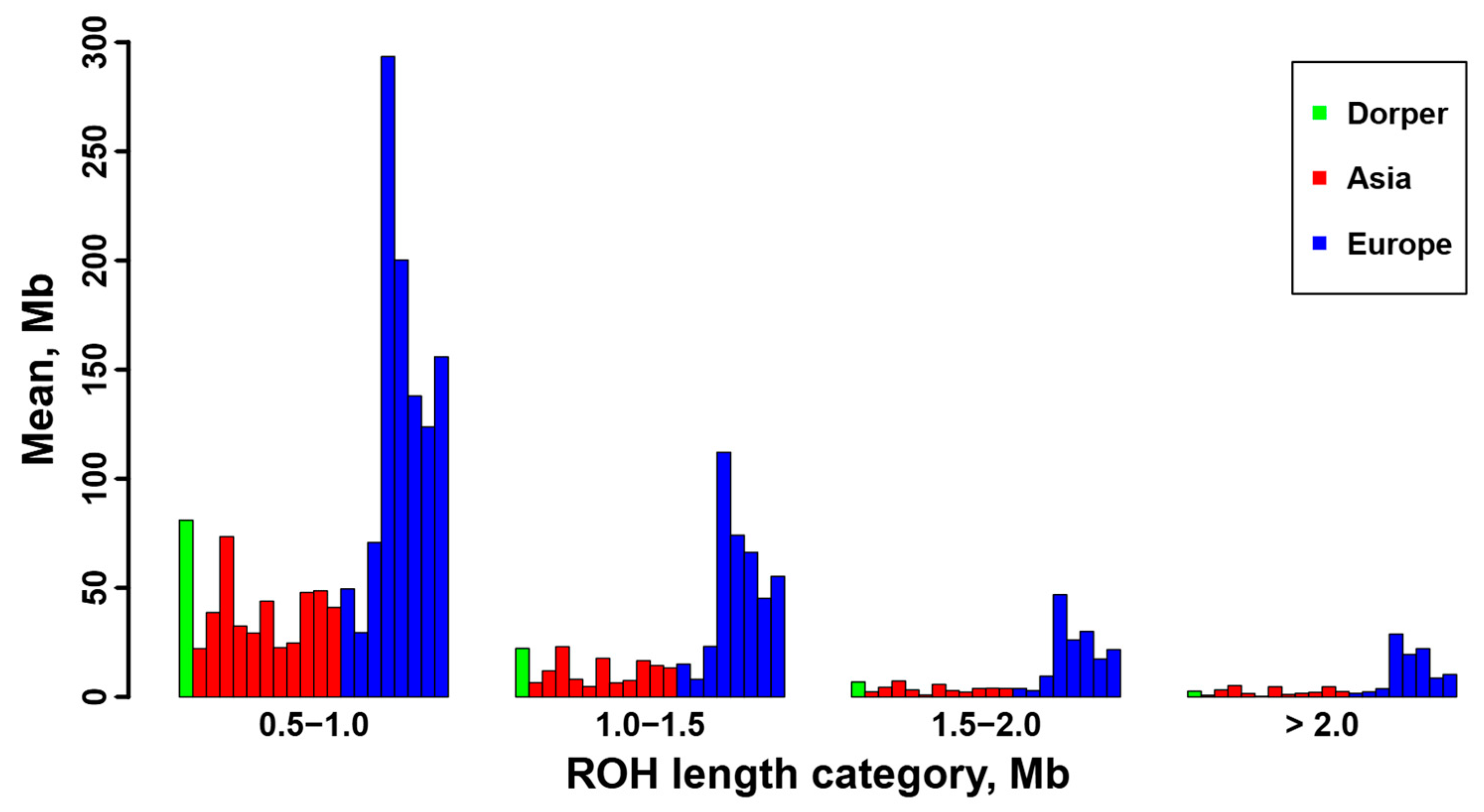
3.3. Genomic Characterization of ROH
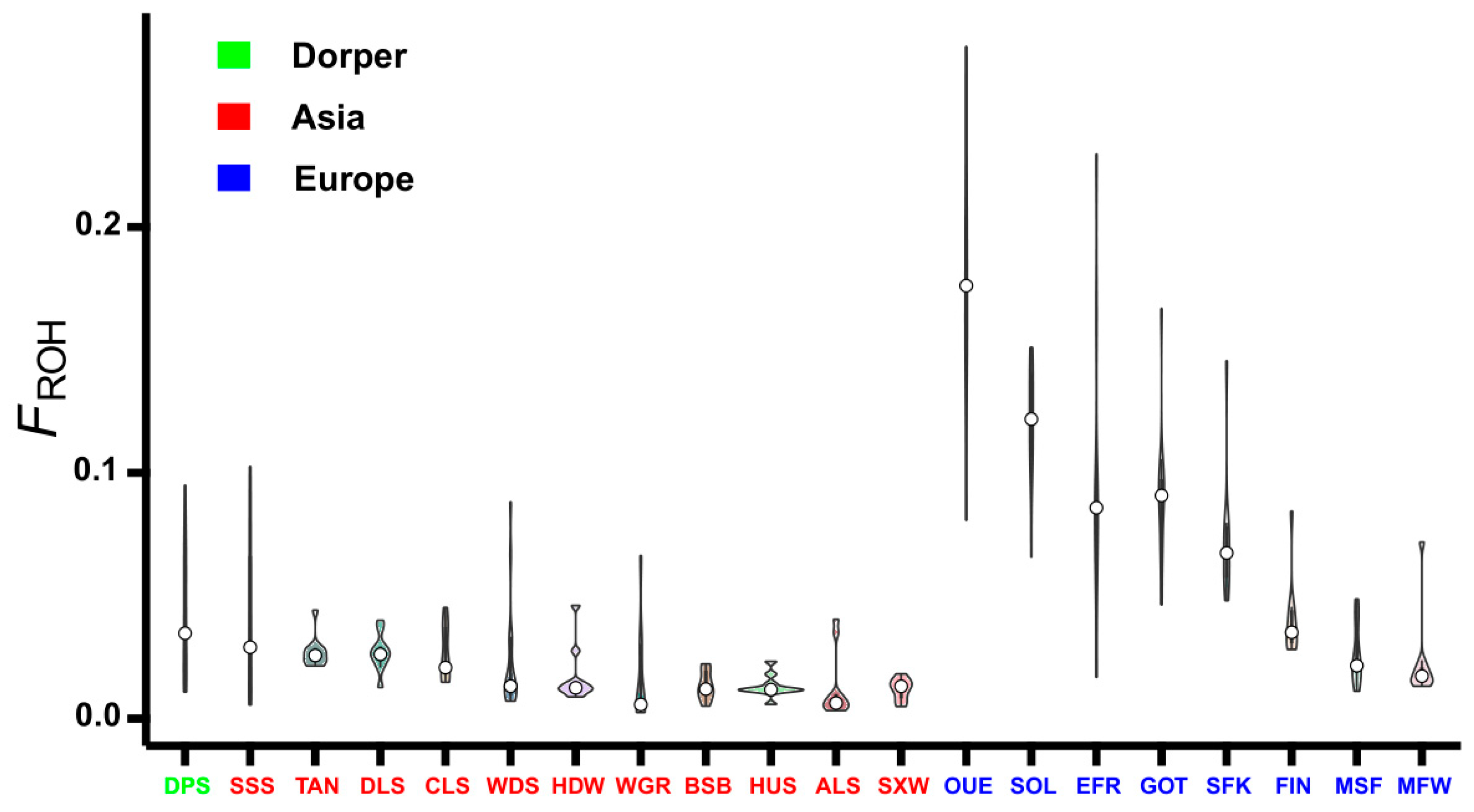
3.4. Inbreeding Coefficient Based on ROH
3.5. ROH Islands and Candidate Genes for Various Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baird, D.; Fairbairn, A.; Jenkins, E.; Martin, L.; Middleton, C.; Pearson, J.; Asouti, E.; Edwards, Y.; Kabukcu, C.; Mustafaoglu, G.; et al. Agricultural origins on the Anatolian plateau. Proc. Natl. Acad. Sci. USA 2018, 115, E3077–E3086. [Google Scholar] [CrossRef]
- Zeder, M.A. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc. Natl. Acad. Sci. USA 2008, 105, 11597–11604. [Google Scholar] [CrossRef]
- Doekes, H.P.; Bijma, P.; Windig, J.J. How Depressing Is Inbreeding? A Meta-Analysis of 30 Years of Research on the Effects of Inbreeding in Livestock. Genes 2021, 12, 926. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. The genetic basis of inbreeding depression. Genet. Res. 1999, 74, 329–340. [Google Scholar] [CrossRef]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef] [PubMed]
- Antonios, S.; Rodríguez-Ramilo, S.T.; Aguilar, I.; Astruc, J.M.; Legarra, A.; Vitezica, Z.G. Genomic and pedigree estimation of inbreeding depression for semen traits in the Basco-Bearnaise dairy sheep breed. J. Dairy Sci. 2021, 104, 3221–3230. [Google Scholar] [CrossRef]
- Tao, L.; He, X.Y.; Wang, X.Y.; Di, R.; Chu, M.X. Litter Size of Sheep (Ovis aries): Inbreeding Depression and Homozygous Regions. Genes 2021, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, M.; Nanaei, H.A.; Javanmard, A.; Esmailizadeh, A. The pattern of runs of homozygosity and genomic inbreeding in world-wide sheep populations. Genomics 2021, 113, 1407–1415. [Google Scholar] [CrossRef]
- Wright, S. Coefficients of inbreeding and relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef]
- Oliehoek, P.A.; Bijma, P. Effects of pedigree errors on the efficiency of conservation decisions. Genet. Sel. Evol. 2009, 41, 9. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Ferencakovic, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Saravanan, K.A.; Panigrahi, M.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B.P. Genome-wide analysis of genetic diversity and selection signatures in three Indian sheep breeds. Livest. Sci. 2021, 243, 104367. [Google Scholar] [CrossRef]
- Caivio-Nasner, S.; López-Herrera, A.; González-Herrera, L.G.; Rincón, J.C. Diversity analysis, runs of homozygosity and genomic inbreeding reveal recent selection in Blanco Orejinegro cattle. J. Anim. Breed. Genet. 2021, 138, 613–627. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Ribani, A.; Moscatelli, G.; Bonacini, M.; Prandi, M.; Mancin, E.; Mantovani, R.; Dall’Olio, S.; Fontanesi, L. Comparative analysis of inbreeding parameters and runs of homozygosity islands in 2 Italian autochthonous cattle breeds mainly raised in the Parmigiano-Reggiano cheese production region. J. Dairy Sci. 2022, 105, 2408–2425. [Google Scholar] [CrossRef]
- Ziegler, T.E.; Molina, A.; Ramón, M.; Sanchez, M.; Muñoz-Mejias, E.; Antonini, A.; Demyda-Peyrás, S. Analysis of the genomic landscape of inbreeding in two divergent groups of Spanish Florida goats. J. Anim. Breed. Genet. 2023, 140, 316–329. [Google Scholar] [CrossRef]
- Xiong, J.K.; Bao, J.J.; Hu, W.P.; Shang, M.Y.; Zhang, L. Whole-genome resequencing reveals genetic diversity and selection characteristics of dairy goat. Front. Genet. 2023, 13, 1044017. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Muñoz, M.; Ribani, A.; Alves, E.; Araújo, J.P.; Bozzi, R.; Candek-Potokar, M.; Charneca, R.; Fernandez, A.; et al. Runs of homozygosity provide a genome landscape picture of inbreeding and genetic history of European autochthonous and commercial pig breeds. Anim. Genet. 2021, 52, 155–170. [Google Scholar] [CrossRef]
- Szmatola, T.; Jasielczuk, I.; Semik-Gurgul, E.; Szyndler-Nedza, M.; Blicharski, T.; Szulc, K.; Skrzypczak, E.; Gurgul, A. Detection of runs of homozygosity in conserved and commercial pig breeds in Poland. J. Anim. Breed. Genet. 2020, 137, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Talebi, R.; Szmatola, T.; Mészáros, G.; Qanbari, S. Runs of Homozygosity in Modern Chicken Revealed by Sequence Data. G3-Genes Genomes Genet. 2020, 10, 4615–4623. [Google Scholar] [CrossRef]
- Wang, H.L.; Wang, Q.; Tan, X.D.; Wang, J.; Zhang, J.; Zheng, M.Q.; Zhao, G.P.; Wen, J. Estimation of genetic variability and identification of regions under selection based on runs of homozygosity in Beijing-You Chickens. Poult. Sci. 2023, 102, 102342. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Zhu, B.; Tang, X.W.; Chen, B.; Liu, M.; Gao, N.; Li, S.; Gu, J.J. Genome-Wide Assessment of Runs of Homozygosity by Whole-Genome Sequencing in Diverse Horse Breeds Worldwide. Genes 2023, 14, 1211. [Google Scholar] [CrossRef] [PubMed]
- Criscione, A.; Mastrangelo, S.; D’Alessandro, E.; Tumino, S.; Di Gerlando, R.; Zumbo, A.; Marletta, D.; Bordonaro, S. Genome-wide survey on three local horse populations with a focus on runs of homozygosity pattern. J. Anim. Breed. Genet. 2022, 139, 540–555. [Google Scholar] [CrossRef]
- Letko, A.; Hédan, B.; Snell, A.; Harris, A.C.; Jagannathan, V.; Andersson, G.; Holst, B.S.; Ostrander, E.A.; Quignon, P.; André, C.; et al. Genomic Diversity and Runs of Homozygosity in Bernese Mountain Dogs. Genes 2023, 14, 650. [Google Scholar] [CrossRef]
- Sams, A.J.; Boyko, A.R. Fine-Scale Resolution of Runs of Homozygosity Reveal Patterns of Inbreeding and Substantial Overlap with Recessive Disease Genotypes in Domestic Dogs. G3-Genes Genomes Genet. 2019, 9, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Absher, D.; Feldman, M.W.; Myers, R.M.; Rosenberg, N.A.; Li, J.Z. Genomic Patterns of Homozygosity in Worldwide Human Populations. Am. J. Hum. Genet. 2012, 91, 275–292. [Google Scholar] [CrossRef]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 2007, 89, 391–403. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.P.; Silva, M.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Shi, L.Y.; Li, Y.; Chen, L.; Garrick, D.; Wang, L.X.; Zhao, F.P. Estimates of genomic inbreeding and identification of candidate regions that differ between Chinese indigenous sheep breeds. J. Anim. Sci. Biotechnol. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.; Shen, M.; Xie, X.L.; Liu, G.J.; Xu, Y.X.; Lv, F.H.; Yang, H.; Yang, Y.L.; Liu, C.B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Xie, X.L.; Wang, D.F.; Zhao, C.; Lv, F.H.; Li, X.; Yang, J.; Yu, J.L.; Shen, M.; Gao, L.; et al. Paternal Origins and Migratory Episodes of Domestic Sheep. Curr. Biol. 2020, 30, 4085–4095. [Google Scholar] [CrossRef]
- Lv, F.H.; Cao, Y.H.; Liu, G.J.; Luo, L.Y.; Lu, R.; Liu, M.J.; Li, W.R.; Zhou, P.; Wang, X.H.; Shen, M.; et al. Whole-Genome Resequencing of Worldwide Wild and Domestic Sheep Elucidates Genetic Diversity, Introgression, and Agronomically Important Loci. Mol. Biol. Evol. 2022, 39, msab353. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 10 April 2024).
- Barbato, M.; Orozco-TerWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Sardina, M.T.; Sottile, G.; Pilla, F.; Portolano, B.; BiOvIta, C. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Zhao, L.M.; Yuan, L.; Li, F.D.; Zhang, X.X.; Tian, H.B.; Ma, Z.W.; Zhang, D.Y.; Zhang, Y.K.; Zhao, Y.; Huang, K.; et al. Whole-genome resequencing of Hu sheep identifies candidate genes associated with agronomic traits. J. Genet. Genom. 2024, 51, 866–876. [Google Scholar] [CrossRef]
- Liu, Q.L.; Sun, Y.X.; Guan, L.J.; Chen, X.N.; Zhou, J.; Liu, P.; Huo, B. Detection of the effect of microvibrational stimulation on human discarded immature oocytes by single-cell transcriptome sequencing technology. J. Assist. Reprod. Genet. 2023, 40, 1773–1781. [Google Scholar] [CrossRef]
- Hatzirodos, N.; Hummitzsch, K.; Irving-Rodgers, H.F.; Breen, J.; Perry, V.E.A.; Anderson, R.A.; Rodgers, R.J. Transcript abundance of stromal and thecal cell related genes during bovine ovarian development. PLoS ONE 2019, 14, e0213575. [Google Scholar] [CrossRef]
- Ietta, F.; Wu, Y.H.; Winter, J.; Xu, L.; Wang, J.X.; Post, M.; Caniggia, I. Dynamic HIF1A regulation during human placental development. Biol. Reprod. 2006, 75, 112–121. [Google Scholar] [CrossRef]
- Zhao, R.X.; Song, N.N.; Ning, X.; Chen, X.H.; Ma, R. ZNF146 regulates cell cycle progression via TFDP1 and DEPDC1B in ovarian cancer cells. Reproduction 2024, 168, e230484. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.P.; Hong, Q.H.; Liu, Y.F.; Ren, C.H.; He, X.Y.; Jiang, Y.T.; Ouyang, Y.N.; Chu, M.X.; Zhang, Z.J. Oviduct Transcriptomic Reveals the Regulation of mRNAs and lncRNAs Related to Goat Prolificacy in the Luteal Phase. Animals 2022, 12, 2823. [Google Scholar] [CrossRef] [PubMed]
- Zang, S.Q.; Yang, X.R.; Ye, J.F.; Mo, X.H.; Zhou, G.B.; Fang, Y. Quantitative phosphoproteomics explain cryopreservation-induced reductions in ram sperm motility. J. Proteom. 2024, 298, 105153. [Google Scholar] [CrossRef]
- Zhao, F.P.; Xie, R.; Fang, L.Z.; Xiang, R.D.; Yuan, Z.H.; Liu, Y.; Wang, L.X. Analysis of 206 whole-genome resequencing reveals selection signatures associated with breed-specific traits in Hu sheep. Evol. Appl. 2024, 17, e13697. [Google Scholar] [CrossRef]
- Yao, G.D.; He, J.H.; Kong, Y.; Zhai, J.; Xu, Y.J.; Yang, G.; Kong, D.Q.; Dong, F.L.; Shi, S.L.; Yang, Q.L.; et al. Transcriptional profiling of long noncoding RNAs and their target transcripts in ovarian cortical tissues from women with normal menstrual cycles and primary ovarian insufficiency. Mol. Reprod. Dev. 2019, 86, 847–861. [Google Scholar] [CrossRef]
- Zhang, W.T.; Jin, M.L.; Lu, Z.K.; Li, T.T.; Wang, H.H.; Yuan, Z.H.; Wei, C.H. Whole Genome Resequencing Reveals Selection Signals Related to Wool Color in Sheep. Animals 2023, 13, 3265. [Google Scholar] [CrossRef]
- Rochus, C.M.; Sunesson, K.W.; Jonas, E.; Mikko, S.; Johansson, A.M. Mutations in ASIP and MC1R: Dominant black and recessive black alleles segregate in native Swedish sheep populations. Anim. Genet. 2019, 50, 712–717. [Google Scholar] [CrossRef]
- Tian, D.H.; Han, B.Y.; Li, X.; Liu, D.H.; Zhou, B.C.; Zhao, C.C.; Zhang, N.; Wang, L.; Pei, Q.B.; Zhao, K. Genetic diversity and selection of Tibetan sheep breeds revealed by whole-genome resequencing. Anim. Biosci. 2023, 36, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.Q.; Li, L.L.; Liu, M.; Wang, L.; Gao, X.X.; Zhou, L.S.; Liu, N.; He, J.N. miR-27a Targeting PIK3R3 Regulates the Proliferation and Apoptosis of Sheep Hair Follicle Stem Cells. Animals 2023, 13, 141. [Google Scholar] [CrossRef]
- Ma, G.W.; Wang, S.Z.; Wang, N.; Li, H.; Yang, H. A Study of Combined Genotype Effects of SHCBP1 on Wool Quality Traits in Chinese Merino. Biochem. Genet. 2023, 61, 551–564. [Google Scholar] [CrossRef]
- Settle, S.H.; Rountree, R.B.; Sinha, A.; Thacker, A.; Higgins, K.; Kingsley, D.M. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev. Biol. 2003, 254, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Lee, E.J.; Jung, S.C.; Lee, J.Y.; Chung, H.W.; Kim, H.L. Genetic variants that affect length/height in infancy/early childhood in Vietnamese-Korean families. J. Hum. Genet. 2010, 55, 681–690. [Google Scholar] [CrossRef]
- Signer-Hasler, H.; Burren, A.; Ammann, P.; Drgemller, C.; Flury, C. Runs of homozygosity and signatures of selection: A comparison among eight local Swiss sheep breeds. Anim. Genet. 2019, 50, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Kominakis, A.; Hager-Theodorides, A.L.; Zoidis, E.; Saridaki, A.; Antonakos, G.; Tsiamis, G. Combined GWAS and ‘guilt by association’-based prioritization analysis identifies functional candidate genes for body size in sheep. Genet. Sel. Evol. 2017, 49, 41. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Song, H.L.; Jiang, Y.F.; Jiang, Y.; Zhang, F.X.; Liu, Y.B.; Shi, Y.; Ding, X.D.; Wang, C.D. A Single-Step Genome Wide Association Study on Body Size Traits Using Imputation-Based Whole-Genome Sequence Data in Yorkshire Pigs. Front. Genet. 2021, 12, 629049. [Google Scholar] [CrossRef]
- Vanvanhossou, S.F.U.; Scheper, C.; Dossa, L.H.; Yin, T.; Brügemann, K.; König, S. A multi-breed GWAS for morphometric traits in four Beninese indigenous cattle breeds reveals loci associated with conformation, carcass and adaptive traits. BMC Genom. 2020, 21, 783. [Google Scholar] [CrossRef]
- Prieur, V.; Clarke, S.M.; Brito, L.F.; McEwan, J.C.; Lee, M.A.; Brauning, R.; Dodds, K.G.; Auvray, B. Estimation of linkage disequilibrium and effective population size in New Zealand sheep using three different methods to create genetic maps. BMC Genet. 2017, 18, 68. [Google Scholar] [CrossRef]
- Peter, C.; Bruford, M.; Perez, T.; Dalamitra, S.; Hewitt, G.; Erhardt, G.; Consortium, E. Genetic diversity and subdivision of 57 European and Middle-Eastern sheep breeds. Anim. Genet. 2007, 38, 37–44. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef]
- Islam, R.; Li, Y.F.; Liu, X.X.; Berihulay, H.; Abied, A.; Gebreselassie, G.; Ma, Q.; Ma, Y.H. Genome-Wide Runs of Homozygosity, Effective Population Size, and Detection of Positive Selection Signatures in Six Chinese Goat Breeds. Genes 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Conrad, L.; Hoerl, J.; Henke, M.; Luick, R.; Schoof, N. Sheep in the Vineyard: Suitability of Different Breeds and Potential Breeding Objectives. Animals 2022, 12, 2575. [Google Scholar] [CrossRef]
- Rochus, C.M.; Jonas, E.; Johansson, A.M. Population structure of five native sheep breeds of Sweden estimated with high density SNP genotypes. BMC Genet. 2020, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.S.; Gao, L.; Xie, X.L.; Ren, Y.L.; Shen, Z.Q.; Wang, F.; Shen, M.; Eypórsdóttir, E.; Hallsson, J.H.; Kiseleva, T.; et al. Genome-Wide Association Analyses Highlight the Potential for Different Genetic Mechanisms for Litter Size Among Sheep Breeds. Front. Genet. 2018, 9, 118. [Google Scholar] [CrossRef]
- Huang, Y.W.; Yuan, C.F.; Zhao, Y.; Li, C.J.; Cao, M.S.; Li, H.B.; Zhao, Z.J.; Sun, A.; Basang, W.D.; Zhu, Y.B.; et al. Identification and Regulatory Network Analysis of Genes Related to Reproductive Performance in the Hypothalamus and Pituitary of Angus Cattle. Genes 2022, 13, 965. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Li, H.L.; Dargahi, D.; Shynlova, O.; Slater, D.; Jones, S.J.M.; Lye, S.J.; Dong, X.S. HoxA13 Regulates Phenotype Regionalization of Human Pregnant Myometrium. J. Clin. Endocrinol. Metab. 2015, 100, E1512–E1522. [Google Scholar] [CrossRef]
- Cunha, G.R.; Robboy, S.J.; Kurita, T.; Isaacson, D.; Shen, J.; Cao, M.; Baskin, L.S. Development of the human female reproductive tract. Differentiation 2018, 103, 46–65. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Trau, H.A.; Masuda, K.; Manley, N.R. Temporal and spatial requirements for Hoxa3 in mouse embryonic development. Dev. Biol. 2016, 415, 33–45. [Google Scholar] [CrossRef]
- Donaldson, I.J.; Amin, S.; Hensman, J.J.; Kutejova, E.; Rattray, M.; Lawrence, N.; Hayes, A.; Ward, C.M.; Bobola, N. Genome-wide occupancy links Hoxa2 to Wnt-β-catenin signaling in mouse embryonic development. Nucleic Acids Res. 2012, 40, 3990–4001. [Google Scholar] [CrossRef]
- Lakhssassi, K.; Sarto, M.P.; Lahoz, B.; Alabart, J.L.; Folch, J.; Serrano, M.; Calvo, J.H. Blood transcriptome of Rasa Aragonesa rams with different sexual behavior phenotype reveals CRYL1 and SORCS2 as genes associated with this trait. J. Anim. Sci. 2023, 101, skad098. [Google Scholar] [CrossRef]
- Wang, W.N.; Chen, S.J.; Qiao, L.Y.; Zhang, S.Y.; Liu, Q.X.; Yang, K.J.; Pan, Y.Y.; Liu, J.H.; Liu, W.Z. Four Markers Useful for the Distinction of Intrauterine Growth Restriction in Sheep. Animals 2023, 13, 3305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Chen, C.; Ahmad, M.J.; Chen, F.; Ding, Z.M.; Yang, S.J.; Chen, Y.W.; Duan, Z.Q.; Liu, M.; Liang, A.X.; et al. WDR62 regulates mouse oocyte meiotic maturation related to p-JNK and H3K9 trimethylation. Int. J. Biochem. Cell Biol. 2022, 144, 106169. [Google Scholar] [CrossRef] [PubMed]
- Ho, U.Y.; Feng, C.W.A.; Yeap, Y.Y.; Bain, A.L.; Wei, Z.; Shohayeb, B.; Reichelt, M.E.; Homer, H.; Khanna, K.K.; Bowles, J.; et al. WDR62 is required for centriole duplication in spermatogenesis and manchette removal in spermiogenesis. Commun. Biol. 2021, 4, 645. [Google Scholar] [CrossRef]
- Eisa, A.A.; De, S.; Detwiler, A.; Gilker, E.; Ignatious, A.C.; Vijayaraghavan, S.; Kline, D. YWHA (14-3-3) protein isoforms and their interactions with CDC25B phosphatase in mouse oogenesis and oocyte maturation. BMC Dev. Biol. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- He, S.G.; Di, J.; Han, B.; Chen, L.; Liu, M.J.; Li, W.R. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino. Animals 2020, 10, 524. [Google Scholar] [CrossRef]
- Qian, Y.; Xiong, Z.; Li, Y.; Kayser, M.; Liu, L.; Liu, F. The effects of Tbx15 and Pax1 on facial and other physical morphology in mice. FASEB bioAdvances 2021, 3, 1011–1019. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breeds | ROH Number (Mean ± SD) | ROH Length, Mb (Mean ± SD) | FROH (Mean ± SD) | FHOM (Mean ± SD) | r(FROH − FHOM) |
|---|---|---|---|---|---|
| DPS | 143.80 ± 93.37 | 0.784 ± 0.297 | 0.0425 ± 0.0288 | 0.0730 ± 0.0523 | 0.913 |
| SXW | 41.50 ± 13.54 | 0.762 ± 0.318 | 0.0119 ± 0.0043 | 0.0271 ± 0.0148 | 0.724 |
| WDS | 71.65 ± 60.10 | 0.809 ± 0.379 | 0.0218 ± 0.0209 | 0.0539 ± 0.0350 | 0.922 |
| SSS | 135.20 ± 116.95 | 0.805 ± 0.356 | 0.0410 ± 0.0378 | 0.0849 ± 0.0698 | 0.995 |
| HDW | 59.00 ± 32.74 | 0.764 ± 0.322 | 0.0170 ± 0.0114 | 0.0411 ± 0.0181 | 0.928 |
| HUS | 49.30 ± 15.87 | 0.708 ± 0.232 | 0.0131 ± 0.0047 | 0.0469 ± 0.0091 | 0.718 |
| TAN | 85.30 ± 19.71 | 0.843 ± 0.387 | 0.0271 ± 0.0065 | 0.0489 ± 0.0111 | 0.893 |
| ALS | 42.30 ± 38.79 | 0.779 ± 0.332 | 0.0124 ± 0.0135 | 0.0154 ± 0.0250 | 0.985 |
| BSB | 45.90 ± 17.74 | 0.785 ± 0.335 | 0.0136 ± 0.0059 | -0.0013 ± 0.0319 | 0.837 |
| CLS | 88.00 ± 35.69 | 0.798 ± 0.309 | 0.0264 ± 0.0113 | 0.0518 ± 0.0229 | 0.988 |
| DLS | 88.20 ± 22.35 | 0.810 ± 0.357 | 0.0269 ± 0.0077 | 0.0407 ± 0.0120 | 0.952 |
| MFW | 75.80 ± 56.17 | 0.798 ± 0.334 | 0.0228 ± 0.0175 | 0.0160 ± 0.0435 | 0.979 |
| MSF | 90.40 ± 44.41 | 0.773 ± 0.294 | 0.0263 ± 0.0127 | 0.0285 ± 0.0381 | 0.956 |
| WGR | 54.00 ± 61.41 | 0.789 ± 0.351 | 0.0161 ± 0.0205 | 0.0355 ± 0.0488 | 0.934 |
| FIN | 130.50 ± 50.17 | 0.821 ± 0.354 | 0.0403 ± 0.0167 | 0.0856 ± 0.0305 | 0.971 |
| OUE | 559.50 ± 159.06 | 0.860 ± 0.392 | 0.1812 ± 0.0552 | 0.3467 ± 0.0689 | 0.713 |
| SOL | 377.60 ± 75.30 | 0.847 ± 0.385 | 0.1204 ± 0.0257 | 0.2167 ± 0.0258 | 0.595 |
| EFR | 281.90 ± 155.49 | 0.909 ± 0.449 | 0.0965 ± 0.0599 | 0.1517 ± 0.0785 | 0.937 |
| SFK | 234.80 ± 81.66 | 0.830 ± 0.355 | 0.0734 ± 0.0275 | 0.1350 ± 0.0503 | 0.956 |
| GOT | 293.80 ± 85.05 | 0.827 ± 0.358 | 0.0915 ± 0.0318 | 0.2005 ± 0.0460 | 0.817 |
| all | 143.81 ± 149.00 | 0.830 ± 0.371 | 0.0450 ± 0.0495 | 0.0835 ± 0.0914 | 0.952 |
| p-value | 2 × 10−16 | 3.23 × 10−14 | 2 × 10−16 | 2 × 10−16 | / |
| Traits | Chromosome | Number of SNPs | Start (bp) | End (bp) | Number of Genes |
|---|---|---|---|---|---|
| Fecundity (European breeds) | 1 | 3568 | 260,516,400 | 261,008,501 | 3 |
| 2 | 7869 | 125,362,089 | 125,929,311 | 2 | |
| 2 | 4788 | 133,972,981 | 134,262,353 | 0 | |
| 2 | 258 | 134,368,535 | 134,393,056 | 0 | |
| 5 | 1810 | 116,823,462 | 117,482,846 | 3 | |
| 10 | 1405 | 28,356,529 | 28,915,246 | 6 | |
| 10 | 3414 | 39,546,231 | 40,127,304 | 1 | |
| 10 | 7324 | 40,690,153 | 41,457,031 | 2 | |
| 10 | 4390 | 45,284,988 | 45,892,572 | 0 | |
| 10 | 125 | 45,893,708 | 45,917,433 | 0 | |
| 10 | 11,555 | 46,066,651 | 47,479,008 | 3 | |
| 11 | 903 | 41,691,551 | 41,766,808 | 4 | |
| 13 | 1736 | 51,484,744 | 52,073,593 | 3 | |
| 13 | 966 | 53,029,769 | 53,185,643 | 1 | |
| 13 | 3722 | 53,185,987 | 53,789,155 | 14 | |
| 13 | 583 | 53,789,304 | 53,954,964 | 2 | |
| 14 | 4564 | 49,339,955 | 49,705,671 | 22 | |
| 14 | 2307 | 52,590,703 | 52,871,923 | 14 | |
| Fecundity (Asian breeds) | 1 | 1264 | 127,784,437 | 128,112,826 | 5 |
| 1 | 1409 | 245,349,451 | 245,668,687 | 0 | |
| 1 | 4564 | 275,364,067 | 276,000,826 | 6 | |
| 2 | 12,148 | 125,310,139 | 126,205,892 | 3 | |
| 2 | 13,140 | 133,294,917 | 134,500,712 | 4 | |
| 2 | 476 | 230,125,601 | 230,160,780 | 1 | |
| 2 | 2246 | 234,992,972 | 235,522,246 | 24 | |
| 4 | 2492 | 75,493,738 | 75,983,090 | 17 | |
| 5 | 5524 | 88,822,803 | 89,607,365 | 2 | |
| 5 | 1030 | 116,948,615 | 117,463,164 | 3 | |
| 6 | 19,452 | 86,306,956 | 87,351,718 | 1 | |
| 6 | 481 | 87,513,615 | 87,568,336 | 0 | |
| 6 | 265 | 87,568,659 | 87,587,663 | 0 | |
| 6 | 4559 | 88,034,854 | 88,359,440 | 1 | |
| 6 | 3883 | 89,450,099 | 90,010,975 | 0 | |
| 6 | 2309 | 129,274,460 | 129,779,428 | 20 | |
| 9 | 2850 | 85,450,258 | 86,050,942 | 4 | |
| 10 | 9390 | 8,565,265 | 9,194,043 | 1 | |
| 10 | 1349 | 28,371,801 | 28,939,707 | 6 | |
| 10 | 516 | 37,585,363 | 37,879,321 | 8 | |
| 10 | 223 | 37,884,095 | 37,921,937 | 2 | |
| 10 | 3402 | 39,546,881 | 40,127,304 | 1 | |
| 10 | 5702 | 40,239,681 | 41,017,766 | 2 | |
| 10 | 1980 | 43,965,011 | 44,184,394 | 0 | |
| 10 | 846 | 44,247,494 | 44,336,764 | 0 | |
| 10 | 3548 | 45,367,439 | 45,879,518 | 0 | |
| 10 | 3979 | 46,456,617 | 46,999,992 | 2 | |
| 10 | 656 | 47,185,534 | 47,220,162 | 0 | |
| 12 | 2981 | 83,917,395 | 84,495,501 | 20 | |
| 13 | 2951 | 51,181,396 | 52,061,687 | 3 | |
| 13 | 1826 | 54,104,798 | 54,613,939 | 28 | |
| 13 | 2676 | 55,938,389 | 56,804,834 | 41 | |
| 20 | 1692 | 54,679,439 | 55,249,641 | 6 | |
| Wool | 2 | 7568 | 133,087,142 | 133,777,850 | 5 |
| 3 | 4451 | 138,495,186 | 138,938,836 | 4 | |
| 3 | 287 | 138,939,171 | 138,994,723 | 0 | |
| 3 | 1226 | 138,995,024 | 139,160,139 | 3 | |
| 4 | 2784 | 75,441,569 | 75,983,090 | 18 | |
| 4 | 4650 | 110,595,183 | 111,105,973 | 3 | |
| 7 | 2608 | 52,547,867 | 53,015,509 | 7 | |
| 7 | 2653 | 91,870,692 | 92,062,931 | 3 | |
| 7 | 4261 | 92,063,098 | 92,595,740 | 16 | |
| 8 | 9502 | 24,759,801 | 25,746,266 | 4 | |
| 8 | 4306 | 26,532,538 | 27,109,398 | 0 | |
| 8 | 357 | 27,973,982 | 28,000,808 | 0 | |
| 9 | 7038 | 11,737,256 | 12,244,991 | 0 | |
| 10 | 17 | 44,162,922 | 44,165,123 | 0 | |
| 10 | 683 | 45,412,636 | 45,501,889 | 0 | |
| 12 | 3758 | 45,584,092 | 46,097,353 | 10 | |
| 12 | 184 | 55,301,863 | 55,390,658 | 3 | |
| 12 | 4656 | 61,885,513 | 62,419,297 | 2 | |
| 13 | 1862 | 54,104,588 | 54,637,805 | 28 | |
| 14 | 1080 | 13,410,344 | 13,668,206 | 1 | |
| 14 | 2632 | 13,894,458 | 14,142,159 | 7 | |
| 14 | 5094 | 14,142,386 | 15,078,390 | 23 | |
| 14 | 9 | 15,078,540 | 15,079,377 | 0 | |
| 14 | 3030 | 15,082,920 | 15,649,580 | 24 | |
| 14 | 491 | 15,652,185 | 15,715,741 | 1 | |
| 15 | 5113 | 1,112,183 | 1,642,914 | 9 | |
| 22 | 4365 | 220,584 | 750,448 | 5 | |
| Body size | 2 | 1254 | 229,295,362 | 229,454,903 | 1 |
| 3 | 2332 | 146,197,096 | 146,567,912 | 12 | |
| 3 | 343 | 146,568,243 | 146,632,643 | 3 | |
| 3 | 992 | 146,714,711 | 146,842,536 | 5 | |
| 3 | 166 | 146,892,649 | 146,927,816 | 1 | |
| 3 | 368 | 146,927,959 | 146,985,136 | 3 | |
| 4 | 2033 | 111,126,770 | 111,331,638 | 4 | |
| 6 | 3612 | 24,489,901 | 24,762,969 | 0 | |
| 6 | 2576 | 47,322,449 | 47,501,103 | 0 | |
| 7 | 7033 | 81,665,952 | 82,295,710 | 2 | |
| 7 | 16 | 99,468,631 | 99,556,337 | 0 | |
| 7 | 1613 | 99,603,078 | 99,736,702 | 0 | |
| 9 | 4933 | 88,768,178 | 89,181,444 | 6 | |
| 10 | 432 | 28,371,264 | 28,503,446 | 3 | |
| 10 | 3005 | 33,282,855 | 33,815,588 | 7 | |
| 11 | 2919 | 28,211,580 | 28,765,976 | 14 | |
| 11 | 4502 | 34,884,456 | 35,764,740 | 55 | |
| 11 | 766 | 35,764,816 | 36,145,259 | 31 | |
| 11 | 667 | 36,145,459 | 36,464,031 | 32 | |
| 11 | 592 | 36,464,533 | 36,602,225 | 11 | |
| 11 | 220 | 37,050,098 | 37,095,273 | 1 | |
| 12 | 8452 | 76,867,182 | 77,507,534 | 11 | |
| 12 | 4075 | 83,728,664 | 84,298,251 | 18 | |
| 13 | 1209 | 23,661,787 | 23,938,741 | 2 | |
| 13 | 5489 | 36,077,863 | 36,646,913 | 6 | |
| 13 | 6143 | 39,564,442 | 40,266,374 | 19 | |
| 13 | 6582 | 40,266,727 | 40,995,074 | 10 | |
| 13 | 2763 | 40,995,703 | 41,369,024 | 3 | |
| 13 | 279 | 41,582,319 | 41,607,838 | 0 | |
| 13 | 3826 | 41,608,135 | 42,271,886 | 8 | |
| 13 | 724 | 45,752,803 | 45,820,785 | 0 | |
| 13 | 2867 | 47,100,995 | 47,377,999 | 0 | |
| 13 | 2406 | 47,378,390 | 47,619,133 | 0 | |
| 13 | 7034 | 47,619,182 | 48,644,663 | 14 | |
| 13 | 1917 | 51,441,819 | 52,093,035 | 3 | |
| 13 | 14 | 52,093,271 | 52,095,647 | 0 | |
| 13 | 1936 | 72,541,472 | 72,890,366 | 12 | |
| 15 | 3608 | 55,819,107 | 56,435,858 | 11 | |
| 18 | 464 | 19,405,282 | 19,474,356 | 0 | |
| 20 | 3362 | 98,697 | 646,780 | 4 | |
| 22 | 2014 | 25,467,538 | 25,974,815 | 18 | |
| 23 | 997 | 30,189,190 | 30,308,004 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, R.; Liu, J.; Ma, X.; Yang, J. Genome-Wide Runs of Homozygosity Reveal Inbreeding Levels and Trait-Associated Candidate Genes in Diverse Sheep Breeds. Genes 2025, 16, 316. https://doi.org/10.3390/genes16030316
Ma R, Liu J, Ma X, Yang J. Genome-Wide Runs of Homozygosity Reveal Inbreeding Levels and Trait-Associated Candidate Genes in Diverse Sheep Breeds. Genes. 2025; 16(3):316. https://doi.org/10.3390/genes16030316
Chicago/Turabian StyleMa, Rui, Jiaxin Liu, Xiao Ma, and Ji Yang. 2025. "Genome-Wide Runs of Homozygosity Reveal Inbreeding Levels and Trait-Associated Candidate Genes in Diverse Sheep Breeds" Genes 16, no. 3: 316. https://doi.org/10.3390/genes16030316
APA StyleMa, R., Liu, J., Ma, X., & Yang, J. (2025). Genome-Wide Runs of Homozygosity Reveal Inbreeding Levels and Trait-Associated Candidate Genes in Diverse Sheep Breeds. Genes, 16(3), 316. https://doi.org/10.3390/genes16030316