

Reprogramming Fibrosis: How Protein PTMs Reshape the IPF Proteome

 , ,
, , 
Abstract
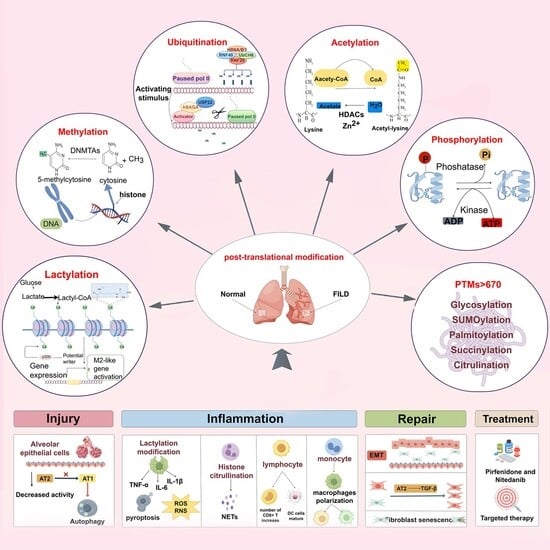
1. Introduction
2. Methods
2.1. Literature Search Strategy
2.2. Screening and Selection of Studies
3. Classification and Functions of PTMs
3.1. Phosphorylation
3.2. Acetylation
3.3. Methylation
3.4. Ubiquitination
3.5. Glycosylation
3.6. SUMOylation
3.7. Lactylation
3.8. Other PTMs
4. Effects of PTMs on Various Stages of IPF
4.1. Injury Stage
4.2. Inflammatory Response Stage
4.2.1. Macrophages
4.2.2. Regulatory T Cells
4.2.3. Other Immune Cells
4.3. Repair Stage
4.3.1. Fibroblasts
4.3.2. LR-MSCs
4.3.3. Myofibroblasts
5. Diagnosis and Treatment of IPF
5.1. Diagnosis of IPF
5.2. Treatment of IPF
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IPF | Idiopathic pulmonary fibrosis |
| PTM | Post-translational modifications |
| NETs | Neutrophil extracellular traps |
| ECM | Extracellular matrix |
| AT2 | Type II alveolar epithelial cells |
| STAT3 | Signal transducer and activator of transcription 3 |
| 4FUT | Fucosyltransferase 4 |
| L1CAM | L1 cell adhesion molecule |
| IFN1 | Type I Interferon |
| NSCLC | Non-small cell lung cancer |
| EMT | Epithelial–Mesenchymal Transition |
| AT1 | Alveolar Type I Cells |
| TPP1 | Telomere protection protein 1 |
| Calpain 1 | Cytosolic calpain 1 |
| ITA3 | Integrin α3β1 |
| AEC2 | Alveolar epithelial cells |
| MoDMs | Monocyte-derived macrophages |
| Treg | Regulatory T cells |
| CS | Cigarette smoke |
| HIF1 | Hypoxia-inducible factor 1 |
| LR-MSCs | Lung-resident mesenchymal stem cells |
| PAD4 | Peptidylarginine deiminase 4 |
| ACP5 | Acid phosphatase 5 |
| GGA | Ginkgolic acid, Glycine–Glycine–Alanine |
| Ythdf1 | YTH N6-methyladenosine RNA binding protein F1 |
| TβR1 | TGF-β type I receptor |
| GLI | Hedgehog/glioma-associated oncogene homolog |
| α-SMA | Smooth muscle α-actinin |
| H3K27ac | Acetylation of histone H3 lysine at position 27 |
| HDAC | Histone deacetylase |
| CTD-ILD | Connective-tissue-disease–associated ILD |
| AGEs | Advanced glycation end-products |
| OTUB1 | Ubiquitin aldehyde-binding protein Otubain1 |
| αPD-L1 | Programmed death ligand 1 |
| ICI | Checkpoint Inhibitors |
| GLP-1R | Glucagon-like peptide-1 receptor |
| Ser209 | Serine 209 |
| FABP5 | Fatty acid binding protein 5 |
| circ_406961 | Cyclic RNA 406961 |
| LMWF | Low-molecular-weight fucoidan |
| ILF2 | Interleukin enhancer-binding factor 2 |
| MAPK8 | Mitogen-activated protein kinase 8 |
| EETs | Epoxyeicosatrienoic acids |
| TPPU | 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea |
| tACPA | Therapeutic anti-citrullinated protein antibodies |
| pSmad2 | Phosphorylated Smad2 |
| MPCs | Mesenchymal progenitor cells |
| ME2 | Mediated phosphorylation of malic enzyme 2 |
| LAR | Lung alveolar regeneration |
| PPF | Progressive Pulmonary Fibrosis |
| METTL3 | methyltransferase-like 3 |
| lnc668 | lncNONMMUT062668.2 |
References
- Hofman, D.E.; Magrì, T.; Moor, C.C.; Richeldi, L.; Wijsenbeek, M.S.; Waseda, Y. Patient-centered care in pulmonary fibrosis: Access, anticipate, and act. Respir. Res. 2024, 25, 395. [Google Scholar] [CrossRef]
- Sun, Z.; He, W.; Meng, H.; Ji, Z.; Qu, J.; Yu, G. Lactate activates ER stress to promote alveolar epithelial cells apoptosis in pulmonary fibrosis. Respir. Res. 2024, 25, 401. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Rana, T.; Jiang, C.; Banerjee, S.; Yi, N.; Zmijewski, J.W.; Liu, G.; Liu, R.M. PAI-1 Regulation of p53 Expression and Senescence in Type II Alveolar Epithelial Cells. Cells 2023, 12, 2008. [Google Scholar] [CrossRef]
- Liu, P.-Y.; Chen, C.-Y.; Lin, Y.-L.; Lin, C.-M.; Tsai, W.-C.; Tsai, Y.-L.; Lin, G.-J.; Chen, Y.-G.; Wang, S.-Y.; Sun, R.-N.; et al. RNF128 regulates neutrophil infiltration and myeloperoxidase functions to prevent acute lung injury. Cell Death Dis. 2023, 14, 369. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Song, C.; Hu, Y.; Dai, M.; Liu, B.; Pan, P. Neutrophil Extracellular Traps Augmented Alveolar Macrophage Pyroptosis via AIM2 Inflammasome Activation in LPS-Induced ALI/ARDS. J. Inflamm. Res. 2021, 14, 4839–4858. [Google Scholar] [CrossRef] [PubMed]
- Agostini, C.; Gurrieri, C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Cao, Y.; Yu, T.; Zhu, Z.; Zhang, Y.; Sun, S.; Li, N.; Gu, C.; Yang, Y. Exploring the landscape of post-translational modification in drug discovery. Pharmacol. Ther. 2025, 265, 108749. [Google Scholar] [PubMed]
- Luo, S.; Martin, B.L.; Senshu, T.; Graves, D.J. Enzymatic deimination of glycogen phosphorylase and a peptide of the phosphorylation site: Identification of modification and roles in phosphorylation and activity. Arch. Biochem. Biophys. 1995, 318, 362–369. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy 2023, 19, 379–387. [Google Scholar]
- Kim, J.; Tadros, B.; Liang, Y.H.; Kim, Y.; Lasagna-Reeves, C.; Sonn, J.Y.; Chung, D.C.; Hyman, B.; Holtzman, D.M.; Zoghbi, H.Y. TYK2 regulates tau levels, phosphorylation and aggregation in a tauopathy mouse model. Nat. Neurosci. 2024, 27, 2417–2429. [Google Scholar] [CrossRef]
- Navarro-Yepes, J.; Anandhan, A.; Bradley, E.; Bohovych, I.; Yarabe, B.; de Jong, A.; Ovaa, H.; Zhou, Y.; Khalimonchuk, O.; Quintanilla-Vega, B.; et al. Inhibition of Protein Ubiquitination by Paraquat and 1-Methyl-4-Phenylpyridinium Impairs Ubiquitin-Dependent Protein Degradation Pathways. Mol. Neurobiol. 2016, 53, 5229–5251. [Google Scholar] [CrossRef]
- Wu, L.; Cheng, Y.; Geng, D.; Fan, Z.; Lin, B.; Zhu, Q.; Li, J.; Qin, W.; Yi, W. O-GlcNAcylation regulates epidermal growth factor receptor intracellular trafficking and signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2107453119. [Google Scholar] [CrossRef] [PubMed]
- Mun, D.G.; Bhat, F.A.; Joshi, N.; Sandoval, L.; Ding, H.; Jain, A.; Peterson, J.A.; Kang, T.; Pujari, G.P.; Tomlinson, J.L.; et al. Diversity of post-translational modifications and cell signaling revealed by single cell and single organelle mass spectrometry. Commun. Biol. 2024, 7, 884. [Google Scholar] [CrossRef]
- Wu, C.J.; Xu, X.; Yuan, D.Y.; Liu, Z.Z.; Tan, L.M.; Su, Y.N.; Li, L.; Chen, S.; He, X.J. Arabidopsis histone acetyltransferase complex coordinates cytoplasmic histone acetylation and nuclear chromatin accessibility. Sci. Adv. 2024, 10, eadp1840. [Google Scholar] [CrossRef]
- Zuo, S.; Zhu, Z.; Liu, Y.; Li, H.; Song, S.; Yin, S. CXCL16 Induces the Progression of Pulmonary Fibrosis through Promoting the Phosphorylation of STAT3. Can. Respir. J. 2019, 2019, 2697376. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, Y.; Zhang, W.; Chen, X. Quercetin ameliorates pulmonary fibrosis by inhibiting SphK1/S1P signaling. Biochem. Cell Biol. 2018, 96, 742–751. [Google Scholar] [PubMed]
- Willette, R.N.; Mangrolia, P.; Pondell, S.M.; Lee, C.Y.W.; Yoo, S.; Rudoltz, M.S.; Cowen, B.R.; Welsch, D.J. Modulation of Oxidative Phosphorylation with IM156 Attenuates Mitochondrial Metabolic Reprogramming and Inhibits Pulmonary Fibrosis. J. Pharmacol. Exp. Ther. 2021, 379, 290–300. [Google Scholar] [CrossRef]
- Hong, X.; Li, N.; Lv, J.; Zhang, Y.; Li, J.; Zhang, J.; Chen, H.F. PTMint database of experimentally verified PTM regulation on protein-protein interaction. Bioinformatics 2023, 39, btac823. [Google Scholar]
- Levene, P.A.; Alsberg, C.L. The Cleavage Products of Vitellin. J. Biol. Chem. 1906, 2, 127–133. [Google Scholar] [CrossRef]
- Duong, V.A.; Lee, H. Bottom-Up Proteomics: Advancements in Sample Preparation. Int. J. Mol. Sci. 2023, 24, 5350. [Google Scholar] [CrossRef]
- Ptacek, J.; Devgan, G.; Michaud, G.; Zhu, H.; Zhu, X.; Fasolo, J.; Guo, H.; Jona, G.; Breitkreutz, A.; Sopko, R.; et al. Global analysis of protein phosphorylation in yeast. Nature 2005, 438, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xie, S.; Cheng, J.; Zhao, Q.; Wu, H.; Jiang, P.; Du, W. AKT1 phosphorylation of cytoplasmic ME2 induces a metabolic switch to glycolysis for tumorigenesis. Nat. Commun. 2024, 15, 686. [Google Scholar] [CrossRef]
- Matsuda, M.; Fujiwara, Y.; Yonezawa, F.; Matsuo, K.; Ishizu, E.; Shimora, H.; Shimizu, S.; Kitatani, K.; Kaibori, Y.; Yamagishi, N.; et al. Cyclin-dependent kinase (CDK) 8 and its paralog CDK19 develop group 2 innate lymphoid cell-related lung fibrosis by activating STAT5. J. Immunol. 2025, vkaf171. [Google Scholar] [CrossRef]
- Knipe, R.S.; Probst, C.K.; Lagares, D.; Franklin, A.; Spinney, J.J.; Brazee, P.L.; Grasberger, P.; Zhang, L.; Black, K.E.; Sakai, N.; et al. The Rho Kinase Isoforms ROCK1 and ROCK2 Each Contribute to the Development of Experimental Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lee, W.; Ravi, D.; Devine, W.; Yong, M.; Diebold, R.B.; Seung, S.A.; Ng, N.W.; Lee, J.; Gupta, A.; et al. Novel Small-Molecule ROCK2 Inhibitor GNS-3595 Attenuates Pulmonary Fibrosis in Preclinical Studies. Am. J. Respir. Cell Mol. Biol. 2024, 71, 430–441. [Google Scholar] [CrossRef]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef]
- Linster, E.; Stephan, I.; Bienvenut, W.V.; Maple-Grødem, J.; Myklebust, L.M.; Huber, M.; Reichelt, M.; Sticht, C.; Møller, S.G.; Meinnel, T.; et al. Downregulation of N-terminal acetylation triggers ABA-mediated drought responses in Arabidopsis. Nat. Commun. 2015, 6, 7640. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, S.; Liu, S.; Qin, D.; Liu, Z.; Wang, L.; Chen, X.; Zhang, L. MSL1 Promotes Liver Regeneration by Driving Phase Separation of STAT3 and Histone H4 and Enhancing Their Acetylation. Adv. Sci. 2023, 10, e2301094. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, P.; Luo, J.; Ding, H.; Cao, H.; He, W.; Zen, K.; Zhou, Y.; Yang, J.; Jiang, L. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021, 12, 847. [Google Scholar] [CrossRef]
- Franker, M.A.; Hoogenraad, C.C. Microtubule-based transport—Basic mechanisms, traffic rules and role in neurological pathogenesis. J. Cell Sci. 2013, 126 Pt 11, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Nowosad, A.; Creff, J.; Jeannot, P.; Culerrier, R.; Codogno, P.; Manenti, S.; Nguyen, L.; Besson, A. p27 controls autophagic vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. Cell Death Dis. 2021, 12, 481. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Poüs, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, Y.; Li, C.; Bi, Y.; Wang, K.; Ye, M.; Li, H. Molecular basis for protein histidine N1-specific methylation of the "His-x-His" motifs by METTL9. Cell Insight 2023, 2, 100090. [Google Scholar] [CrossRef]
- Wang, X.; Xie, H.; Guo, Q.; Cao, D.; Ru, W.; Zhao, S.; Zhu, Z.; Zhang, J.; Pan, W.; Yao, X.; et al. Molecular basis for METTL9-mediated N1-histidine methylation. Cell Discov. 2023, 9, 38. [Google Scholar] [CrossRef]
- Dai, S.; Holt, M.V.; Horton, J.R.; Woodcock, C.B.; Patel, A.; Zhang, X.; Young, N.L.; Wilkinson, A.W.; Cheng, X. Characterization of SETD3 methyltransferase–mediated protein methionine methylation. J. Biol. Chem. 2020, 295, 10901–10910. [Google Scholar] [CrossRef]
- Davydova, E.; Shimazu, T.; Schuhmacher, M.K.; Jakobsson, M.E.; Willemen, H.; Liu, T.; Moen, A.; Ho, A.Y.Y.; Małecki, J.; Schroer, L.; et al. The methyltransferase METTL9 mediates pervasive 1-methylhistidine modification in mammalian proteomes. Nat. Commun. 2021, 12, 891. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, Z.F.; Liu, W.; Kazama, K.; Hu, L.; Sun, X.; Wang, L.; Lee, H.; Lu, L.; Yang, X.F.; et al. PIMT is a novel and potent suppressor of endothelial activation. Elife 2023, 12, e85754. [Google Scholar] [CrossRef]
- Wang, K.; Huang, W.; Chen, R.; Lin, P.; Zhang, T.; Ni, Y.F.; Li, H.; Wu, J.; Sun, X.X.; Geng, J.J.; et al. Di-methylation of CD147-K234 Promotes the Progression of NSCLC by Enhancing Lactate Export. Cell Metab. 2021, 33, 160–173.E6. [Google Scholar] [CrossRef]
- Chen, X.; Huang, M.F.; Fan, D.M.; He, Y.H.; Zhang, W.J.; Ding, J.C.; Peng, B.L.; Pan, X.; Liu, Y.; Du, J.; et al. CARM1 hypermethylates the NuRD chromatin remodeling complex to promote cell cycle gene expression and breast cancer development. Nucleic Acids Res. 2024, 52, 6811–6829. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hyeon, D.Y.; Yoon, S.H.; Jeong, J.H.; Han, S.M.; Jang, J.W.; Nguyen, M.P.; Chi, X.Z.; An, S.; Hyun, K.G.; et al. RUNX3 methylation drives hypoxia-induced cell proliferation and antiapoptosis in early tumorigenesis. Cell Death Differ. 2021, 28, 1251–1269. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, G.; Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Choi, K.M.; Haak, A.J.; Aravamudhan, A.; Roden, A.C.; Prakash, Y.S.; et al. CBX5/G9a/H3K9me-mediated gene repression is essential to fibroblast activation during lung fibrosis. JCI Insight 2019, 5, e127111. [Google Scholar] [CrossRef] [PubMed]
- Jbeli, A.H.; Yang, L.; Xia, H.; Gilbertsen, A.J.; Bitterman, P.B.; Henke, C.A. Brg1/PRMT5 nuclear complex epigenetically regulates FOXO1 in IPF mesenchymal progenitor cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2024, 326, L344–L352. [Google Scholar] [CrossRef]
- Rowbotham, S.P.; Pessina, P.; Garcia-de-Alba, C.; Jensen, J.; Nguyen, Y.; Yoon, J.; Li, J.; Wong, I.G.; Fahey, C.; Moye, A.L.; et al. Age-associated H3K9me2 loss alters the regenerative equilibrium between murine lung alveolar and bronchiolar progenitors. Dev. Cell 2023, 58, 2974–2991.E6. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, H.; Wang, Y.; Liu, W.; Li, G.; Li, D.; Wu, W.; Wu, Y.; Zhang, Z.; Ji, Y.; et al. Noscapine derivative 428 suppresses ferroptosis through targeting GPX4. Redox Biol. 2025, 83, 103635. [Google Scholar] [CrossRef]
- Hung, C.H.; Wu, S.Y.; Yao, C.D.; Yeh, H.H.; Lin, C.C.; Chu, C.Y.; Huang, T.Y.; Shen, M.R.; Lin, C.H.; Su, W.C. Defective N-glycosylation of IL6 induces metastasis and tyrosine kinase inhibitor resistance in lung cancer. Nat. Commun. 2024, 15, 7885. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; He, Z.; Li, Z.; Wang, Y.; Wu, N.; Sun, H.; Zhou, Z.; Hu, Q.; Cong, X. Lactylation: The novel histone modification influence on gene expression, protein function, and disease. Clin. Epigenetics 2024, 16, 72. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.B.; Jiang, H.C.; Li, W.; Li, M.K.; Yang, X.Y.; Cai, Y.Y.; Xue, K.K.; Gou, Y.S.; Liu, X.Y.; et al. LMO7 drives profibrotic fibroblast polarization and pulmonary fibrosis in mice through TGF-β signalling. Acta Pharmacol. Sin. 2025, 46, 1930–1945. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Y.; Feng, J. Identification and validation of CDC20 and ITCH as ubiquitination related biomarker in idiopathic pulmonary fibrosis. Hereditas 2025, 162, 50. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, W.; Han, Y.; Oyang, L.; Lin, J.; Shen, M.; Wang, J.; et al. Altered glycosylation in cancer: Molecular functions and therapeutic potential. Cancer Commun 2024, 44, 1316–1336. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J. Protein glycosylation. Curr. Biol. 2019, 29, R229–R231. [Google Scholar] [CrossRef]
- Sharma, P.; Zhang, X.; Ly, K.; Kim, J.H.; Wan, Q.; Kim, J.; Lou, M.; Kain, L.; Teyton, L.; Winau, F. Hyperglycosylation of prosaposin in tumor dendritic cells drives immune escape. Science 2024, 383, 190–200. [Google Scholar] [CrossRef]
- Liu, Q.; Adhikari, E.; Lester, D.K.; Fang, B.; Johnson, J.O.; Tian, Y.; Mockabee-Macias, A.T.; Izumi, V.; Guzman, K.M.; White, M.G.; et al. Androgen drives melanoma invasiveness and metastatic spread by inducing tumorigenic fucosylation. Nat. Commun. 2024, 15, 1148. [Google Scholar] [CrossRef]
- Arai, J.; Hayakawa, Y.; Tateno, H.; Murakami, K.; Hayashi, T.; Hata, M.; Matsushita, Y.; Kinoshita, H.; Abe, S.; Kurokawa, K.; et al. Impaired Glycosylation of Gastric Mucins Drives Gastric Tumorigenesis and Serves as a Novel Therapeutic Target. Gastroenterology 2024, 167, 505–521.e19. [Google Scholar] [CrossRef]
- Di Bacco, A.; Gill, G. SUMO-specific proteases and the cell cycle. An essential role for SENP5 in cell proliferation. Cell Cycle 2006, 5, 2310–2313. [Google Scholar] [CrossRef]
- Dai, W.; Xie, S.; Chen, C.; Choi, B.H. Ras sumoylation in cell signaling and transformation. Semin. Cancer Biol. 2021, 76, 301–309. [Google Scholar] [CrossRef]
- Demel, U.M.; Böger, M.; Yousefian, S.; Grunert, C.; Zhang, L.; Hotz, P.W.; Gottschlich, A.; Köse, H.; Isaakidis, K.; Vonficht, D.; et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J. Clin. Investig. 2022, 132, e152383. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xia, Q.; Mao, M.; Zhou, H.; Zheng, L.; Wang, Y.; Zeng, Z.; Yan, L.; Zhao, Y.; Shi, J. Annexin-A1 SUMOylation regulates microglial polarization after cerebral ischemia by modulating IKKα stability via selective autophagy. Sci. Adv. 2021, 7, eabc5539. [Google Scholar] [CrossRef] [PubMed]
- Lightcap, E.S.; Yu, P.; Grossman, S.; Song, K.; Khattar, M.; Xega, K.; He, X.; Gavin, J.M.; Imaichi, H.; Garnsey, J.J.; et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci. Transl. Med. 2021, 13, eaba7791. [Google Scholar] [CrossRef] [PubMed]
- Diao, X.; Guo, C.; Zheng, H.; Zhao, K.; Luo, Y.; An, M.; Lin, Y.; Chen, J.; Li, Y.; Li, Y.; et al. SUMOylation-triggered ALIX activation modulates extracellular vesicles circTLCD4-RWDD3 to promote lymphatic metastasis of non-small cell lung cancer. Signal Transduct. Target. Ther. 2023, 8, 426. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, J.; Wang, Y.; Zhao, W.; Wang, W.; Cui, J.; Yang, J.; Yue, Y.; Zhang, S.; Chu, M.; et al. A proteomic atlas of ligand-receptor interactions at the ovine maternal-fetal interface reveals the role of histone lactylation in uterine remodeling. J. Biol. Chem. 2022, 298, 101456. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Hagihara, H.; Shoji, H.; Otabi, H.; Toyoda, A.; Katoh, K.; Namihira, M.; Miyakawa, T. Protein lactylation induced by neural excitation. Cell Rep. 2021, 37, 109820. [Google Scholar] [CrossRef]
- Li, L.; Chen, K.; Wang, T.; Wu, Y.; Xing, G.; Chen, M.; Hao, Z.; Zhang, C.; Zhang, J.; Ma, B.; et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat. Metab. 2020, 2, 882–892. [Google Scholar] [CrossRef]
- Mo, Y.; Han, Y.; Chen, Y.; Fu, C.; Li, Q.; Liu, Z.; Xiao, M.; Xu, B. ZDHHC20 mediated S-palmitoylation of fatty acid synthase (FASN) promotes hepatocarcinogenesis. Mol. Cancer 2024, 23, 274. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, J.; Yang, Y.; Shan, H.; Hou, S.; Fang, H.; Ma, M.; Chen, Z.; Tan, L.; Xu, D. A palmitoylation-depalmitoylation relay spatiotemporally controls GSDMD activation in pyroptosis. Nat. Cell Biol. 2024, 26, 757–769. [Google Scholar] [CrossRef]
- He, Y.; Li, S.; Jiang, L.; Wu, K.; Chen, S.; Su, L.; Liu, C.; Liu, P.; Luo, W.; Zhong, S.; et al. Palmitic Acid Accelerates Endothelial Cell Injury and Cardiovascular Dysfunction via Palmitoylation of PKM2. Adv. Sci. 2025, 12, e2412895. [Google Scholar] [CrossRef]
- Linder, M.E.; Deschenes, R.J. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef]
- Nadolski, M.J.; Linder, M.E. Protein lipidation. Febs J. 2007, 274, 5202–5210. [Google Scholar] [CrossRef]
- Nie, L.; Fei, C.; Fan, Y.; Dang, F.; Zhao, Z.; Zhu, T.; Wu, X.; Dai, T.; Balasubramanian, A.; Pan, J.; et al. Consecutive palmitoylation and phosphorylation orchestrates NLRP3 membrane trafficking and inflammasome activation. Mol. Cell 2024, 84, 3336–3353.E7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zhen, J.; Liu, X.; Guo, J.; Li, D.; Xie, J.; Xie, L. Protein post-translational modification by lysine succinylation: Biochemistry, biological implications, and therapeutic opportunities. Genes. Dis. 2023, 10, 1242–1262. [Google Scholar] [CrossRef]
- Ma, W.; Sun, Y.; Yan, R.; Zhang, P.; Shen, S.; Lu, H.; Zhou, Z.; Jiang, Z.; Ye, L.; Mao, Q.; et al. OXCT1 functions as a succinyltransferase, contributing to hepatocellular carcinoma via succinylating LACTB. Mol. Cell 2024, 84, 538–551.E7. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Xu, J.; Wang, L.; Yuan, Y.; Luo, R.; Gan, M.; Wang, K.; Zhao, T.; Wang, Y.; Han, T.; et al. SUCLG2 Regulates Mitochondrial Dysfunction through Succinylation in Lung Adenocarcinoma. Adv. Sci. 2023, 10, e2303535. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Dong, S.; Kawada, A.; Kojima, T.; Chavanas, S.; Méchin, M.C.; Adoue, V.; Serre, G.; Simon, M.; Takahara, H. Transcriptional regulation of peptidylarginine deiminase expression in human keratinocytes. J. Dermatol. Sci. 2009, 53, 2–9. [Google Scholar] [CrossRef]
- Rebak, A.S.; Hendriks, I.A.; Elsborg, J.D.; Buch-Larsen, S.C.; Nielsen, C.H.; Terslev, L.; Kirsch, R.; Damgaard, D.; Doncheva, N.T.; Lennartsson, C.; et al. A quantitative and site-specific atlas of the citrullinome reveals widespread existence of citrullination and insights into PADI4 substrates. Nat. Struct. Mol. Biol. 2024, 31, 977–995. [Google Scholar] [CrossRef]
- Watanabe, S.; Markov, N.S.; Lu, Z.; Piseaux Aillon, R.; Soberanes, S.; Runyan, C.E.; Ren, Z.; Grant, R.A.; Maciel, M.; Abdala-Valencia, H.; et al. Resetting proteostasis with ISRIB promotes epithelial differentiation to attenuate pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2101100118. [Google Scholar] [CrossRef]
- Franzén, L.; Olsson Lindvall, M.; Hühn, M.; Ptasinski, V.; Setyo, L.; Keith, B.P.; Collin, A.; Oag, S.; Volckaert, T.; Borde, A.; et al. Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis. Nat. Genet. 2024, 56, 1725–1736. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, D.; Bin, E.; Li, J.; Jiang, K.; Lv, T.; Mao, X.; Wang, F.; Dai, H.; Tang, N. Enhanced glycolysis-mediated energy production in alveolar stem cells is required for alveolar regeneration. Cell Stem Cell 2023, 30, 1028–1042.E7. [Google Scholar] [CrossRef]
- Lv, X.; Liu, S.; Liu, C.; Li, Y.; Zhang, T.; Qi, J.; Li, K.; Hua, F.; Cui, B.; Zhang, X.; et al. TRIB3 promotes pulmonary fibrosis through inhibiting SLUG degradation by physically interacting with MDM2. Acta Pharm. Sin. B 2023, 13, 1631–1647. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, R.; Li, G.; Wang, Z.; Liu, J.; Liang, Y.; Liu, J.P. FBW7 Mediates Senescence and Pulmonary Fibrosis through Telomere Uncapping. Cell Metab. 2020, 32, 860–877.E9. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Cheng, H.; Li, X.; Qiu, Y.; Zhang, Y.; Chang, Y.; Fu, J.; Shen, M.; Xu, X.; Feng, D.; et al. Abnormal mitochondrial iron metabolism damages alveolar type II epithelial cells involved in bleomycin-induced pulmonary fibrosis. Theranostics 2024, 14, 2687–2705. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Yu, L.; Bian, X.; Zhang, C.; Wu, Z.; Huang, N.; Yang, J.; Jin, W.; Feng, Z.; Li, D.; Huo, X.; et al. Ginkgolic acid improves bleomycin-induced pulmonary fibrosis by inhibiting SMAD4 SUMOylation. Oxid. Med. Cell Longev. 2022, 2022, 8002566. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, N.; Margadant, C.; Kevelam, S.H.; Lilien, M.R.; Oosterveld, M.J.; Kreft, M.; van Eerde, A.M.; Pfundt, R.; Terhal, P.A.; van der Zwaag, B.; et al. Gain of glycosylation in integrin α3 causes lung disease and nephrotic syndrome. J. Clin. Investig. 2012, 122, 4375–4387. [Google Scholar] [CrossRef]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef]
- Fu, Y.; Pajulas, A.; Wang, J.; Zhou, B.; Cannon, A.; Cheung, C.C.L.; Zhang, J.; Zhou, H.; Fisher, A.J.; Omstead, D.T.; et al. Mouse pulmonary interstitial macrophages mediate the pro-tumorigenic effects of IL-9. Nat. Commun. 2022, 13, 3811. [Google Scholar] [CrossRef]
- Lv, J.; Gao, H.; Ma, J.; Liu, J.; Tian, Y.; Yang, C.; Li, M.; Zhao, Y.; Li, Z.; Zhang, X.; et al. Dynamic atlas of immune cells reveals multiple functional features of macrophages associated with progression of pulmonary fibrosis. Front. Immunol. 2023, 14, 1230266. [Google Scholar] [CrossRef]
- Liu, S.S.; Lv, X.X.; Liu, C.; Qi, J.; Li, Y.X.; Wei, X.P.; Li, K.; Hua, F.; Cui, B.; Zhang, X.W.; et al. Targeting Degradation of the Transcription Factor C/EBPβ Reduces Lung Fibrosis by Restoring Activity of the Ubiquitin-Editing Enzyme A20 in Macrophages. Immunity 2019, 51, 522–534.E7. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Li, H.; Dai, X.; Zhou, J.; Wang, Y.; Zhang, S.; Guo, J.; Shen, L.; Yan, H.; Jiang, H. Mitochondrial dynamics in pulmonary disease: Implications for the potential therapeutics. J. Cell Physiol. 2024, 239, e31370. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, P.; Dong, M.; Zhang, Y.; Lu, S.; Chen, M.; Zhou, H.; Lin, N.; Jiang, H.; Wang, Y. SLC15A3 plays a crucial role in pulmonary fibrosis by regulating macrophage oxidative stress. Cell Death Differ. 2024, 31, 417–430. [Google Scholar] [CrossRef]
- Mehrotra, P.; Maschalidi, S.; Boeckaerts, L.; Maueröder, C.; Tixeira, R.; Pinney, J.; Burgoa Cardás, J.; Sukhov, V.; Incik, Y.; Anderson, C.J.; et al. Oxylipins and metabolites from pyroptotic cells act as promoters of tissue repair. Nature 2024, 631, 207–215. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Xie, Y.; Tan, Q.; Zhou, C.; Gu, P.; Zhang, Y.; Yang, S.; Yin, H.; Shang, B.; Yao, Y.; et al. Glycolytic reprogramming governs crystalline silica-induced pyroptosis and inflammation through promoting lactylation modification. Ecotoxicol. Environ. Saf. 2024, 283, 116952. [Google Scholar] [CrossRef]
- Ouyang, B.; Deng, L.; Yang, F.; Shi, H.; Wang, N.; Tang, W.; Huang, X.; Zhou, Y.; Yu, H.; Wei, Y.; et al. Albumin-based formononetin nanomedicines for lung injury and fibrosis therapy via blocking macrophage pyroptosis. Mater. Today Bio 2023, 20, 100643. [Google Scholar] [CrossRef]
- Boveda-Ruiz, D.; D’Alessandro-Gabazza, C.N.; Toda, M.; Takagi, T.; Naito, M.; Matsushima, Y.; Matsumoto, T.; Kobayashi, T.; Gil-Bernabe, P.; Chelakkot-Govindalayathil, A.L.; et al. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology 2013, 218, 245–254. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R.; et al. Control of TH17/Treg Balance by Hypoxia-Inducible Factor 1. Cell 2011, 146, 772–784. [Google Scholar]
- Chen, Z.; Barbi, J.; Bu, S.; Yang, H.-Y.; Li, Z.; Gao, Y.; Jinasena, D.; Fu, J.; Lin, F.; Chen, C.; et al. The Ubiquitin Ligase Stub1 Negatively Modulates Regulatory T Cell Suppressive Activity by Promoting Degradation of the Transcription Factor Foxp3. Immunity 2013, 39, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Li, B.; Song, X.; Bembas, K.; Zhang, G.; Katsumata, M.; Saouaf, S.J.; Wang, Q.; Hancock, W.W.; Shen, Y.; et al. TGF-β and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc. Natl. Acad. Sci. USA 2008, 105, 14023–14027. [Google Scholar] [CrossRef] [PubMed]
- van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.; Beekman, J.M.; van Beekum, O.; Brenkman, A.B.; Hijnen, D.J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Pan, X.; Zhan, Y.; Zhou, W.; Peng, S.; Chen, C.; Zhang, M.; Lan, R.; Wu, J.; et al. Glycyrrhetinic Acid Mitigates Radiation-Induced Pulmonary Fibrosis via Inhibiting the Secretion of TGF-β1 by Treg Cells. Int. J. Radiat. Oncol. Biol. Phys. 2024, 118, 218–230. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Ikari, J.; Anazawa, R.; Tanaka, N.; Katsumata, Y.; Shimada, A.; Suzuki, E.; Tatsumi, K. PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am. J. Respir. Cell Mol. Biol. 2020, 63, 806–818. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Günes Günsel, G.; Conlon, T.M.; Jeridi, A.; Kim, R.; Ertüz, Z.; Lang, N.J.; Ansari, M.; Novikova, M.; Jiang, D.; Strunz, M.; et al. The arginine methyltransferase PRMT7 promotes extravasation of monocytes resulting in tissue injury in COPD. Nat. Commun. 2022, 13, 1303. [Google Scholar] [CrossRef]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef]
- Sun, T.; Huang, Z.; Liang, W.C.; Yin, J.; Lin, W.Y.; Wu, J.; Vernes, J.M.; Lutman, J.; Caplazi, P.; Jeet, S.; et al. TGFβ2 and TGFβ3 isoforms drive fibrotic disease pathogenesis. Sci. Transl. Med. 2021, 13, eabe0407. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Q.; Yu, J.; Zhou, Q.; Deng, Y.; Liu, J.; Zhang, L.; Xu, Y.; Xiong, W.; Wang, Y. Tartrate-resistant acid phosphatase 5 promotes pulmonary fibrosis by modulating β-catenin signaling. Nat. Commun. 2022, 13, 114. [Google Scholar] [CrossRef]
- Li, J.; Zeng, G.; Zhang, Z.; Wang, Y.; Shao, M.; Li, C.; Lu, Z.; Zhao, Y.; Zhang, F.; Ding, W. Urban airborne PM(2.5) induces pulmonary fibrosis through triggering glycolysis and subsequent modification of histone lactylation in macrophages. Ecotoxicol. Environ. Saf. 2024, 273, 116162. [Google Scholar] [CrossRef]
- Cui, H.; Xie, N.; Banerjee, S.; Ge, J.; Jiang, D.; Dey, T.; Matthews, Q.L.; Liu, R.M.; Liu, G. Lung Myofibroblasts Promote Macrophage Profibrotic Activity through Lactate-induced Histone Lactylation. Am. J. Respir. Cell Mol. Biol. 2021, 64, 115–125. [Google Scholar] [CrossRef]
- Wang, P.; Xie, D.; Xiao, T.; Cheng, C.; Wang, D.; Sun, J.; Wu, M.; Yang, Y.; Zhang, A.; Liu, Q. H3K18 lactylation promotes the progression of arsenite-related idiopathic pulmonary fibrosis via YTHDF1/m6A/NREP. J. Hazard. Mater. 2024, 461, 132582. [Google Scholar] [CrossRef]
- Zheng, J.; Du, Y.; Shao, W.; Li, J.; Zhao, P.; Zhang, Q. Effective-compounds of Jinshui Huanxian Formula acts as an SRC inhibitor to inhibit HK2-mediated H3K18 lactation and improve pulmonary fibrosis. Phytomedicine 2025, 140, 156628. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Zhang, J.; Liu, B.; Liu, W.; Cao, G.; Li, R.; Li, H.; Zhai, N.; Song, X.; et al. YTHDC1 phase separation drives the nuclear export of m(6)A-modified lncNONMMUT062668.2 through the transport complex SRSF3-ALYREF-XPO5 to aggravate pulmonary fibrosis. Cell Death Dis. 2025, 16, 279. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, L.; Zhang, Z.; Tang, H. The emerging roles of SUMOylation in pulmonary diseases. Mol. Med. 2023, 29, 119. [Google Scholar] [CrossRef] [PubMed]
- Louzada, R.A.; Corre, R.; Ameziane El Hassani, R.; Meziani, L.; Jaillet, M.; Cazes, A.; Crestani, B.; Deutsch, E.; Dupuy, C. NADPH oxidase DUOX1 sustains TGF-β1 signalling and promotes lung fibrosis. Eur. Respir. J. 2021, 57, 1901949. [Google Scholar] [CrossRef]
- Li, F.; Gong, X.; Yang, B. Geranylgeranylacetone ameliorated ischemia/reperfusion induced-blood brain barrier breakdown through HSP70-dependent anti-apoptosis effect. Am. J. Transl. Res. 2021, 13, 102–114. [Google Scholar]
- Zhou, R.; Jin, C.; Jiao, L.; Zhang, S.; Tian, M.; Liu, J.; Yang, S.; Yao, W.; Zhou, F. Geranylgeranylacetone, an inducer of heat shock protein 70, attenuates pulmonary fibrosis via inhibiting NF-κB/NOX4/ROS signalling pathway in vitro and in vivo. Chem. Biol. Interact. 2023, 382, 110603. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Calver, J.F.; Parmar, N.R.; Harris, G.; Lithgo, R.M.; Stylianou, P.; Zetterberg, F.R.; Gooptu, B.; Mackinnon, A.C.; Carr, S.B.; Borthwick, L.A.; et al. Defining the mechanism of galectin-3-mediated TGF-β1 activation and its role in lung fibrosis. J. Biol. Chem. 2024, 300, 107300. [Google Scholar] [CrossRef]
- Gao, S.; Li, X.; Jiang, Q.; Liang, Q.; Zhang, F.; Li, S.; Zhang, R.; Luan, J.; Zhu, J.; Gu, X.; et al. PKM2 promotes pulmonary fibrosis by stabilizing TGF-β1 receptor I and enhancing TGF-β1 signaling. Sci. Adv. 2022, 8, eabo0987. [Google Scholar] [CrossRef]
- Lee, C.M.; Jarrell, Z.R.; Lee, H.Y.; Singer, G.; Tran, V.T.; Orr, M.; Jones, D.P.; Go, Y.M. Protein S-palmitoylation enhances profibrotic signaling in response to cadmium. Toxicol. Appl. Pharmacol. 2024, 483, 116806. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra247. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Jarrell, Z.R.; Liang, Y.; Ryan Smith, M.; Orr, M.L.; Marts, L.; Go, Y.M.; Jones, D.P. Vanadium pentoxide induced oxidative stress and cellular senescence in human lung fibroblasts. Redox Biol. 2022, 55, 102409. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A., Jr.; Hogaboam, C.M. Quercetin Enhances Ligand-induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef]
- Ramos, C.; Montaño, M.; García-Alvarez, J.; Ruiz, V.; Uhal, B.D.; Selman, M.; Pardo, A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am. J. Respir. Cell Mol. Biol. 2001, 24, 591–598. [Google Scholar] [CrossRef]
- Enomoto, Y.; Katsura, H.; Fujimura, T.; Ogata, A.; Baba, S.; Yamaoka, A.; Kihara, M.; Abe, T.; Nishimura, O.; Kadota, M.; et al. Autocrine TGF-β-positive feedback in profibrotic AT2-lineage cells plays a crucial role in non-inflammatory lung fibrogenesis. Nat. Commun. 2023, 14, 4956. [Google Scholar] [CrossRef] [PubMed]
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-β and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 22, 6107. [Google Scholar] [CrossRef]
- Yang, S.; Liu, P.; Jiang, Y.; Wang, Z.; Dai, H.; Wang, C. Therapeutic Applications of Mesenchymal Stem Cells in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2021, 9, 639657. [Google Scholar] [CrossRef]
- Sun, W.; Liu, X.; Yang, X.; Jing, X.; Duan, C.; Yang, G.; Wu, C.; Huang, H.; Luo, Q.; Xia, S.; et al. SENP1 regulates the transformation of lung resident mesenchymal stem cells and is associated with idiopathic pulmonary fibrosis progression. Cell Commun. Signal 2022, 20, 104. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Chen, H.; Li, H.; Xu, P.; Liu, B.; Zhang, Q.; Lv, C.; Song, X. ATF3 -activated accelerating effect of LINC00941/lncIAPF on fibroblast-to-myofibroblast differentiation by blocking autophagy depending on ELAVL1/HuR in pulmonary fibrosis. Autophagy 2022, 18, 2636–2655. [Google Scholar] [CrossRef]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta 2007, 1773, 1572–1582. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, Y.; Sun, Y.; Song, M.; Pang, J.; Wang, M.; Zhang, Z.; Yang, P.; Chen, Y.; Qi, X.; et al. Proteome, Lysine Acetylome, and Succinylome Identify Posttranslational Modification of STAT1 as a Novel Drug Target in Silicosis. Mol. Cell Proteom. 2024, 23, 100770. [Google Scholar] [CrossRef]
- Cabrera Cesar, E.; Lopez-Lopez, L.; Lara, E.; Hidalgo-San Juan, M.V.; Parrado Romero, C.; Palencia, J.; Martín-Montañez, E.; Garcia-Fernandez, M. Serum Biomarkers in Differential Diagnosis of Idiopathic Pulmonary Fibrosis and Connective Tissue Disease-Associated Interstitial Lung Disease. J. Clin. Med. 2021, 10, 3167. [Google Scholar] [CrossRef]
- Mo, C.; Li, H.; Yan, M.; Xu, S.; Wu, J.; Li, J.; Yang, X.; Li, Y.; Yang, J.; Su, X.; et al. Dopaminylation of endothelial TPI1 suppresses ferroptotic angiocrine signals to promote lung regeneration over fibrosis. Cell Metab. 2024, 36, 1839–1857.E12. [Google Scholar] [CrossRef]
- Liu, S.S.; Liu, C.; Lv, X.X.; Cui, B.; Yan, J.; Li, Y.X.; Li, K.; Hua, F.; Zhang, X.W.; Yu, J.J.; et al. The chemokine CCL1 triggers an AMFR-SPRY1 pathway that promotes differentiation of lung fibroblasts into myofibroblasts and drives pulmonary fibrosis. Immunity 2021, 54, 2042–2056.E8. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Milara, J.; Abdeldjebbar, Y.; Gubara, S.; Jones, C.; Bueno-Beti, C.; Chepurko, E.; Kohlbrenner, E.; Katz, M.G.; Tarzami, S.; et al. AAV1.SERCA2a Gene Therapy Reverses Pulmonary Fibrosis by Blocking the STAT3/FOXM1 Pathway and Promoting the SNON/SKI Axis. Mol. Ther. 2020, 28, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Duerr, J.; Leitz, D.H.W.; Szczygiel, M.; Dvornikov, D.; Fraumann, S.G.; Kreutz, C.; Zadora, P.K.; Seyhan Agircan, A.; Konietzke, P.; Engelmann, T.A.; et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 2020, 11, 2012. [Google Scholar] [CrossRef]
- Tu, J.; Xu, H.; Ma, L.; Li, C.; Qin, W.; Chen, X.; Yi, M.; Sun, L.; Liu, B.; Yuan, X. Nintedanib enhances the efficacy of PD-L1 blockade by upregulating MHC-I and PD-L1 expression in tumor cells. Theranostics 2022, 12, 747–766. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Q.; Zhou, H.; Jin, L.; Liu, C.; Yang, M.; Zhao, X.; Ding, W.; Xie, W.; Kong, H. GLP-1R activation attenuates the progression of pulmonary fibrosis via disrupting NLRP3 inflammasome/PFKFB3-driven glycolysis interaction and histone lactylation. J. Transl. Med. 2024, 22, 954. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Z.; Liu, J.; Song, M.; Zhang, T.; Chen, Y.; Hu, H.; Yang, P.; Li, B.; Song, X.; et al. Gefitinib and fostamatinib target EGFR and SYK to attenuate silicosis: A multi-omics study with drug exploration. Signal Transduct. Target. Ther. 2022, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Shi, H.; Zhang, D.; Wang, C.; Zhao, F.; Li, L.; Xu, Z.; Jiang, J.; Li, J. Nebulized inhalation of LPAE-HDAC10 inhibits acetylation-mediated ROS/NF-κB pathway for silicosis treatment. J. Control. Release 2023, 364, 618–631. [Google Scholar] [CrossRef]
- Mei, Q.; Yang, Z.; Xiang, Z.; Zuo, H.; Zhou, Z.; Dong, X.; Zhang, L.; Song, W.; Wang, Y.; Hu, Q.; et al. Pharmacological inhibition of MDM4 alleviates pulmonary fibrosis. Theranostics 2023, 13, 2787–2799. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Zhong, W.J.; Liu, Y.B.; Duan, J.X.; Jiang, N.; Yang, H.H.; Ma, S.C.; Jin, L.; Hong, J.R.; Zhou, Y.; et al. EETs alleviate alveolar epithelial cell senescence by inhibiting endoplasmic reticulum stress through the Trim25/Keap1/Nrf2 axis. Redox Biol. 2023, 63, 102765. [Google Scholar] [CrossRef]
- Walker, N.M.; Ibuki, Y.; McLinden, A.P.; Misumi, K.; Mitchell, D.C.; Kleer, G.G.; Lock, A.M.; Vittal, R.; Sonenberg, N.; Garner, A.L.; et al. MNK-driven eIF4E phosphorylation regulates the fibrogenic transformation of mesenchymal cells and chronic lung allograft dysfunction. J. Clin. Investig. 2024, 134, e168393. [Google Scholar] [CrossRef]
- Lei, Q.; Yu, Z.; Li, H.; Cheng, J.; Wang, Y. Fatty acid-binding protein 5 aggravates pulmonary artery fibrosis in pulmonary hypertension secondary to left heart disease via activating wnt/β-catenin pathway. J. Adv. Res. 2022, 40, 197–206. [Google Scholar] [CrossRef]
- Mair, K.M.; MacLean, M.R.; Morecroft, I.; Dempsie, Y.; Palmer, T.M. Novel interactions between the 5-HT transporter, 5-HT1B receptors and Rho kinase in vivo and in pulmonary fibroblasts. Br. J. Pharmacol. 2008, 155, 606–616. [Google Scholar] [CrossRef]
- Xiang, F.L.; Fang, M.; Yutzey, K.E. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Li, Z.; Wang, J.; Geng, L.; Yue, Y.; Deng, Z.; Wang, Q.; Zhang, Q. Low molecular weight fucoidan attenuating pulmonary fibrosis by relieving inflammatory reaction and progression of epithelial-mesenchymal transition. Carbohydr. Polym. 2021, 273, 118567. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef]
- Le, T.T.; Karmouty-Quintana, H.; Melicoff, E.; Le, T.T.; Weng, T.; Chen, N.Y.; Pedroza, M.; Zhou, Y.; Davies, J.; Philip, K.; et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J. Immunol. 2014, 193, 3755–3768. [Google Scholar] [CrossRef] [PubMed]
- Pedroza, M.; Le, T.T.; Lewis, K.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. Faseb J. 2016, 30, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10, eaar8356. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, X.; Nan, A.; Zhang, N.; Chen, L.; Zhou, H.; Zhang, H.; Qiu, M.; Zhu, J.; Ling, Y.; et al. Circular RNA 406961 interacts with ILF2 to regulate PM(2.5)-induced inflammatory responses in human bronchial epithelial cells via activation of STAT3/JNK pathways. Environ. Int. 2020, 141, 105755. [Google Scholar] [CrossRef]
- Keum, H.; Kim, J.; Yoo, D.; Kim, T.W.; Seo, C.; Kim, D.; Jon, S. Biomimetic lipid Nanocomplexes incorporating STAT3-inhibiting peptides effectively infiltrate the lung barrier and ameliorate pulmonary fibrosis. J. Control. Release 2021, 332, 160–170. [Google Scholar] [CrossRef]
- Wang, W.; Bale, S.; Wei, J.; Yalavarthi, B.; Bhattacharyya, D.; Yan, J.J.; Abdala-Valencia, H.; Xu, D.; Sun, H.; Marangoni, R.G.; et al. Fibroblast A20 governs fibrosis susceptibility and its repression by DREAM promotes fibrosis in multiple organs. Nat. Commun. 2022, 13, 6358. [Google Scholar] [CrossRef]
- Jarrell, Z.R.; Lee, C.M.; Kim, K.H.; He, X.; Smith, M.R.; Raha, J.R.; Bhatnagasr, N.; Orr, M.; Kang, S.M.; Chen, Y.; et al. Metabolic reprograming and increased inflammation by cadmium exposure following early-life respiratory syncytial virus infection-the involvement of protein S-palmitoylation. Toxicol. Sci. 2023, 197, 186–196. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef]
- Castellanos-Moreira, R.; Rodríguez-García, S.C.; Haro, I.; Sanmarti, R. Response to: ‘Autoantibodies and interstitial lung disease in rheumatoid arthritis: Towards a ‘mix-and-match’ approach’ by Alunno et al. Ann. Rheum. Dis. 2022, 81, e54. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Modification | Function | References |
|---|---|---|---|
| Phosphorylation | Ser, Thr and Tyr | Energy and metabolism regulation, ECM deposition, fibroblast-to-myofibroblast transformation | [23,24,25,26,27,28,29,30] |
| Acetylation | Lys | Energy, autophagy and cell-cycle regulation | [31,32,33,34,35,36,37] |
| Methylation | Lys, Arg, His, Asp | Cell cycle, cell microenvironment, self-renewal of MPCs, fibroblast-to-myofibroblast conversion | [38,39,40,41,42,43,44,45,46,47,48] |
| Ubiquitination | Lys | Macromolecule stability, ferroptosis, collagen deposition, EMT | [49,52,53,54,55] |
| Glycosylation | Asp, Ser and Thr | Cell ferroptosis [49] and interaction | [50,56,57,58,59,60,61] |
| SUMOylation | Lys | Inflammatory factor release | [62,63,64,65,66,67] |
| Lactylation | Lys | Metabolic reprogramming | [51,68,69,70,71] |
| Palmitoylation | Cys | Energy and metabolism regulation, NLRP3 transfer, pyroptosis | [72,73,74,75,76,77] |
| Succinylation | Lys | Mitochondrial dysfunction, energy supply | [78,79,80] |
| Citrullination | Arg | Inflammatory response | [81,82] |
| Stage | Related Cells | Effects on Cells | Post-Translational Modification Types | References |
|---|---|---|---|---|
| Injury | AT2 1,2,3,5 | AT2 to AT1 conversion, AT2 cell self-renewal | Ubiquitination | [85,86,87] |
| AT2 cell activity | Ubiquitination | [88,89] | ||
| Inflammation | Macrophage 1,5 | Phenotype transformation | Ubiquitination | [92,93,94] |
| Oxidative stress | Phosphorylation | [96,97,98] | ||
| Pyroptosis | Lactylation | [99,100,101] | ||
| Regulatory T cells 1,2,5 | Secretion of TGF-β | Ubiquitination, acetylation, phosphorylation and glycosylation | [102,103,104,105,106,107] | |
| Other immune cells 1,2,5 | NETosis | Citrullination | [108,109] | |
| MoDMs Accumulation | Methylation | [110,111] | ||
| Repair | Fibroblasts 1,2,3,4 | TGF-β axis | Phosphorylation, acetylation, lactylation, glycosylation, ubiquitination, palmitoylation and SUMOylation | [112,113,114,115,116,117,118,119,120,121,122,123,124,125] |
| Fibroblast senescence | S-glutathionylation | [130,131,132] | ||
| Interactions with AT2 cells | Lactylation | [119,135] | ||
| Lung-resident mesenchymal stem cells 1,2 | Transformation into myofibroblasts | SUMOylation | [136,137,138] | |
| Myofibroblast 1,2,5 | The number and function of myofibroblasts | Acetylation | [139,140,141,142] | |
| Epithelial cells 1,2,6 | EMT | Phosphorylation, SUMOylation and Glycosylation | [90,91,143] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Kong, W.; Zhang, H.; Wei, X.; Yi, J.; Wang, M.; Jin, S.; Yu, D. Reprogramming Fibrosis: How Protein PTMs Reshape the IPF Proteome. Genes 2025, 16, 1392. https://doi.org/10.3390/genes16111392
Li Y, Kong W, Zhang H, Wei X, Yi J, Wang M, Jin S, Yu D. Reprogramming Fibrosis: How Protein PTMs Reshape the IPF Proteome. Genes. 2025; 16(11):1392. https://doi.org/10.3390/genes16111392
Chicago/Turabian StyleLi, Yunze, Wei Kong, Hanqi Zhang, Xinfeng Wei, Junxuan Yi, Mingwei Wang, Shunzi Jin, and Duo Yu. 2025. "Reprogramming Fibrosis: How Protein PTMs Reshape the IPF Proteome" Genes 16, no. 11: 1392. https://doi.org/10.3390/genes16111392
APA StyleLi, Y., Kong, W., Zhang, H., Wei, X., Yi, J., Wang, M., Jin, S., & Yu, D. (2025). Reprogramming Fibrosis: How Protein PTMs Reshape the IPF Proteome. Genes, 16(11), 1392. https://doi.org/10.3390/genes16111392