
Genetic Heterogeneity in Cowpea Genotypes (Vigna unguiculata L. Walp) Using DArTseq (GBS)-Derived Single Nucleotide Polymorphisms

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Sampling Procedure
2.2. DNA Extraction
2.3. DArTseq Analysis
2.4. SNP Calling
2.5. Data Analysis
3. Results
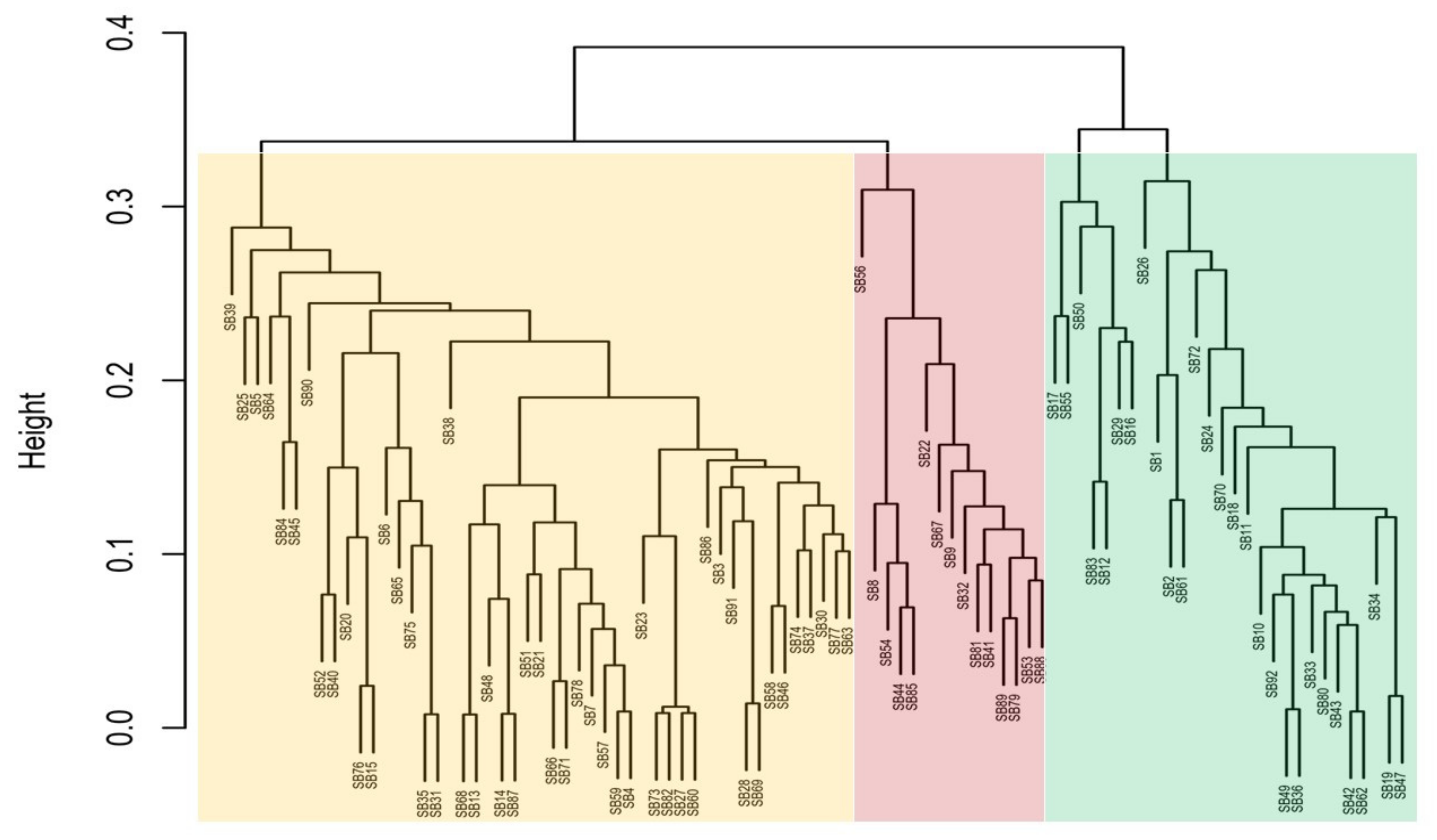
3.1. Cluster Dendrogram (UPGMA)
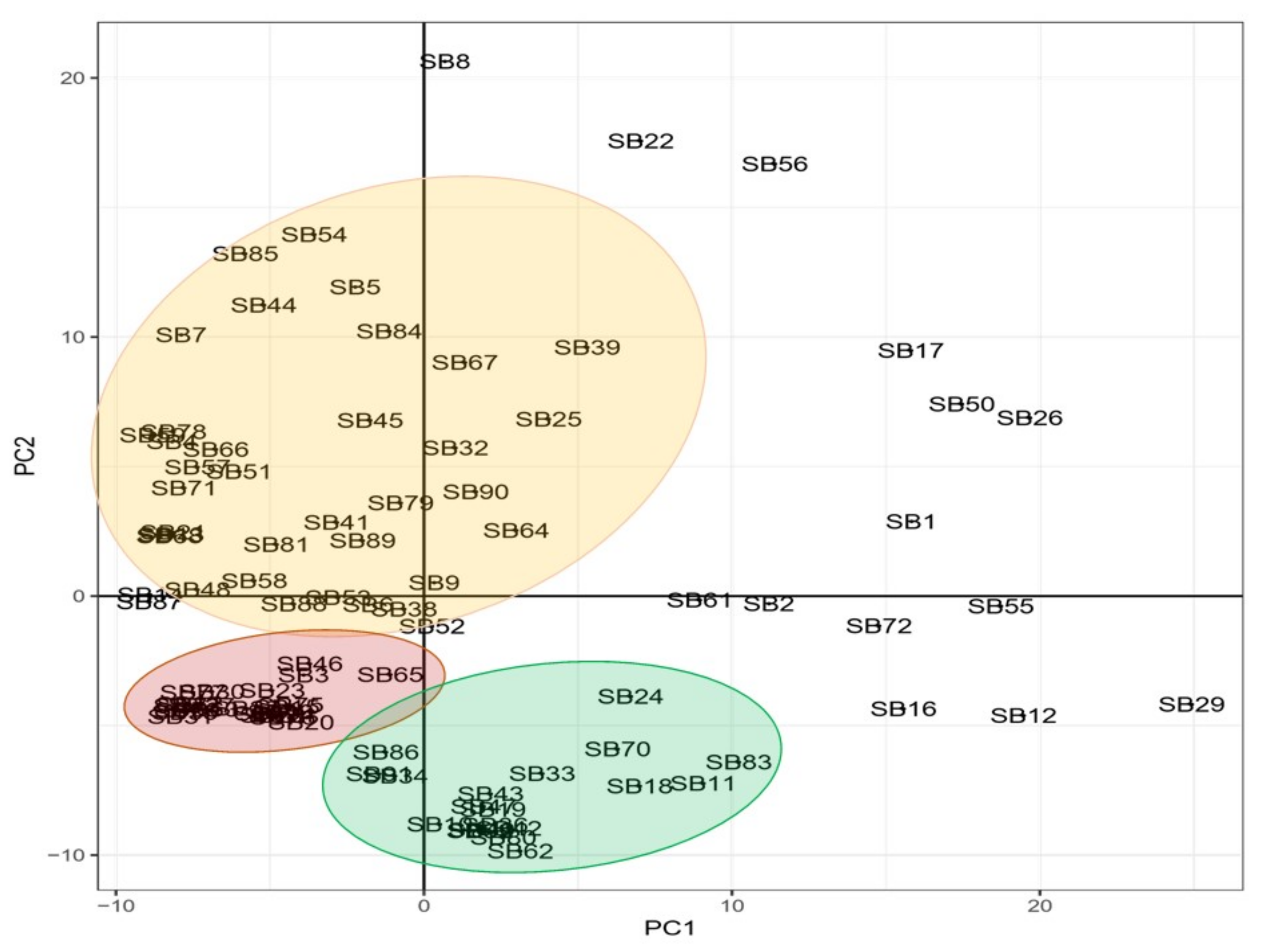
3.2. Principal Component Analysis
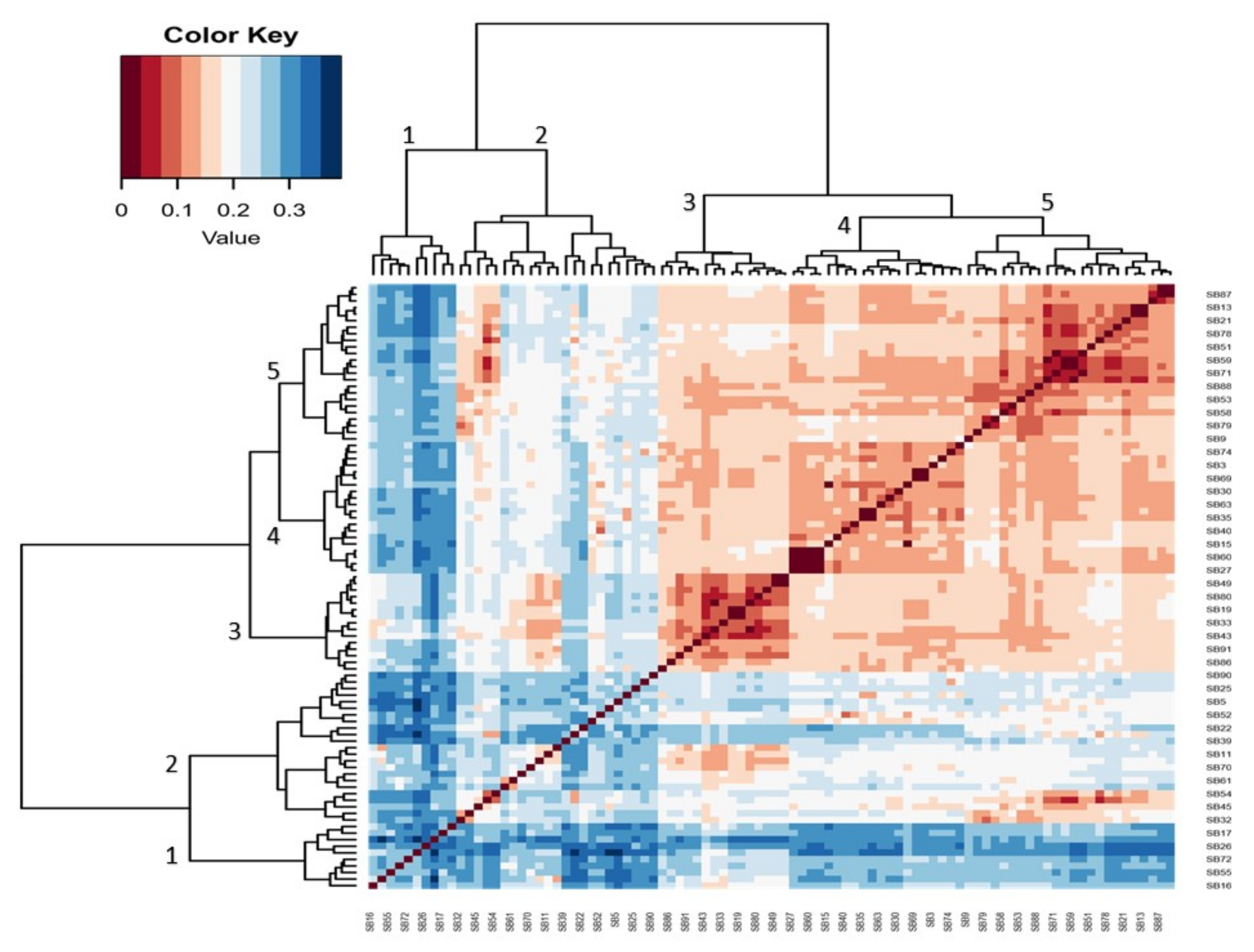
3.3. Hierarchical Heatmap
3.4. Heterogeneity Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Da Silva, D.O.M.; Santos, C.A.F.; Boiteux, L.S. Adaptability and stability parameters of total seed yield and protein content in cowpea (Vigna unguiculata) genotypes subjected to semi-arid conditions. Aust. J. Crop Sci. 2016, 10, 1164–1169. [Google Scholar] [CrossRef]
- Nkoana, D.K.; Gerrano, A.S.; Gwata, E.T. Agronomic performance and genetic variability of cowpea (Vigna unguiculata) accessions. Legume Res. Int. J. 2019, 42, 757–762. [Google Scholar] [CrossRef]
- FAO. World Food and Agriculture—Statistical Yearbook 2021; FAO: Rome, Italy, 2022; ISBN 978-9-5-134332-6. [Google Scholar] [CrossRef]
- Mohamed, H.I.; El-Beltagi, H.S.; Yousef, H.N.; Fawzi, E.M. Biological and Pharmacological Activities of Cowpea (Vigna unguiculata L.) and Its Bioactive Components. In Cowpea: Research Progress and Management Challenges; Nikolic, B.A., Ed.; Nova Science Publishers: New York, NY, USA, 2018; Chapter 4; ISBN 978-1-53614-282-2. [Google Scholar]
- Mohammed, S.B.; Mohammad, I.F.; Pangirayi, T.B.; Vernon, G.; Dzidzienyo, D.K.; Umar, M.L.; Umar, S. Farmers’ knowledge, perception, and use of phosphorus fertilization for cowpea production in Northern Guinea Savannah of Nigeria. Heliyon 2020, 6, e05207. [Google Scholar] [CrossRef] [PubMed]
- Boukar, O.; Belko, N.; Chamarth, S.; Togola, A.; Batieno, J.; Owusu, E.; Haruna, M.; Diallo, S.; Umar, M.L.; Olufajo, O.; et al. Cowpea (Vigna unguiculata): Genetics, genomics, and breeding. Plant Breed. 2018, 138, 415–424. [Google Scholar] [CrossRef]
- Xiong, H.; Shi, A.; Mou, B.; Qin, J.; Motes, D.; Lu, W.; Ma, J.; Weng, Y.; Yang, W.; Wu, D. Genetic diversity and population structure of cowpea (Vigna unguiculata L. Walp). PLoS ONE 2016, 11, e0160941. [Google Scholar] [CrossRef]
- Egea, L.A.; Merida-Garcia, R.; Kilian, A.; Hernandez, P.; Dorado, G. Assessment of genetic diversity and structure of large garlic (Allium sativum) germplasm bank, by Diversity Arrays Technology “Genotyping-by-Sequencing” platform (DArTseq). Front. Genet. 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Kilian, A.; Sanewski, G.; Ko, L. The application of DArTseq technology to pineapple. Int. Soc. Hortic. Sci. 2016, 1111, 181–188. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Pojskić, N. iMAF—Index of Major Allele Frequency. Genet. Appl. 2018, 2, 78–81. [Google Scholar] [CrossRef]
- Malik, S.R.; Shabbir, G.; Zubir, M.; Iqbal, S.M.; Ali, A. Genetic diversity of morpho-genetic traits in Desi Chickpea (Cicer arietinum). Int. J. Agric. Biol. 2014, 16, 956–960. [Google Scholar]
- Nilah, K.; Adak, M.S. Associations of some characters with grain yield in chickpea (Cicer arietinum L.). Pak. J. Bot. 2012, 44, 267–272. [Google Scholar]
- Shete, S.; Tiwari, H.; Elston, R.C. On estimating the heterozygosity and polymorphism information content value. Theor. Popul. Biol. 2000, 57, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mao, W.; Zhang, G.; Wu, F. Development and characterization of Polymorphic EST-SSR and Genomic SSR Markers for Tibetan Annual Wild Barley. PLoS ONE 2014, 9, e94881. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Brock, J.; Dyer, J.M.; Kutchan, T.; Schachtman, D.; Augustin, M.; Ge, Y.; Fahlgren, N.; Abdel-Haleem, H. Genetic Diversity and Population Structure of a Camelina sativa Spring Panel. Front. Plant Sci. 2019, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Gail, M.H.; Weinberg, C.R.; Carroll, R.J.; Chung, C.C.; Wang, Z.; Chanock, S.J.; Fraumeni, J.F.; Chatterjee, N. Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc. Natl. Acad. Sci. USA 2011, 108, 18026–18031. [Google Scholar] [CrossRef] [PubMed]
- Shahsavarani, H.; Rahimi-Mianji, G. Analysis of genetic diversity and estimation of inbreeding coefficient within Caspian horse population using microsatellite markers. Afr. J. Biotechnol. 2010, 9, 293–299. [Google Scholar]
- Huynh, O.A.; Hofinger, B.J.; Beshir, M.M.; Jankowicz-Cieslak, J.; Guo, H.; Forster, B.P.; Till, B.J. Do-it-yourself molecular biology for plant breeding: Low-cost tools for developing countries. In Proceedings of the Plant Genetics and Breeding Technologies, Vienna, Austria, 18–20 February 2013; pp. 33–36. [Google Scholar]
- Melville, J.; Haines, M.L.; Boysen, K.; Hodkinson, L.; Kilian, A.; Smith Date, K.L.; Potvin, D.A.; Parris, K.M. Identifying hybridization and admixture using SNPs: Application of the DArTseq platform in phylogeographic research on vertebrates. R. Soc. Open Sci. 2017, 4, 161061. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Vikram, P.; Singh, R.P.; Kilian, A.; Carling, J.; Song, J.; Burgueno-Ferreira, J.A.; Bhavani, S.; Huerta-Espino, J.; Payne, T.; et al. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genom. 2015, 16, 216. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Genotype Sample Code | Genotype Name | Origin | Genotype Sample Code | Genotype Name | Origin | Genotype Sample Code | Genotype Name | Origin |
|---|---|---|---|---|---|---|---|---|
| SB1 | TVU 14190 | Nigeria | SB32 | RV 351 | South Africa | SB63 | ARC 039 | South Africa |
| SB2 | RV 503 | South Africa | SB33 | 98K-503-1 | Nigeria | SB64 | ARC 022 | South Africa |
| SB3 | TVU 2095 | Nigeria | SB34 | RV 126 | South Africa | SB65 | ARC 029 | South Africa |
| SB4 | BECHUANA WHITE | South Africa | SB35 | TVU 11424 | Nigeria | SB66 | AGRINAWA | South Africa |
| SB5 | TVU 3416 | Nigeria | SB36 | RV 447 | South Africa | SB67 | RV 343 | South Africa |
| SB6 | PAN 311 | South Africa | SB37 | RV 558 | South Africa | SB68 | ARC 012 | South Africa |
| SB7 | RV 204 | South Africa | SB38 | TVU 11986 | Nigeria | SB69 | ARC 020 | South Africa |
| SB8 | RV 194 | South Africa | SB39 | TVU 13778 | Nigeria | SB70 | RV 327 | South Africa |
| SB9 | RV 438 | South Africa | SB40 | 98D-1399 | Nigeria | SB71 | ARC 040 | South Africa |
| SB10 | 95K-589-2 | Nigeria | SB41 | RV 341 | South Africa | SB72 | ORELU | Nigeria |
| SB11 | RV 416 | South Africa | SB42 | RV 440 | South Africa | SB73 | RV 315 | South Africa |
| SB12 | RV 442 | South Africa | SB43 | OLOYIN | Nigeria | SB74 | RV 417 | South Africa |
| SB13 | RV 207 | South Africa | SB44 | RV 202 | South Africa | SB75 | ARC 037 | South Africa |
| SB14 | ARC 045 | South Africa | SB45 | ENCORE 349 | South Africa | SB76 | RV 439 | South Africa |
| SB15 | ARC 024 | South Africa | SB46 | DR SAUNDERS | South Africa | SB77 | ARC 009 | South Africa |
| SB16 | RV 498 | South Africa | SB47 | RV 568 | South Africa | SB78 | ARC 004 | South Africa |
| SB17 | RV 403 | South Africa | SB48 | RV 554 | South Africa | SB79 | RV 352 | South Africa |
| SB18 | 98K-476-8 | Nigeria | SB49 | RV 446 | South Africa | SB80 | RV 457 | South Africa |
| SB19 | IT93K-1294 | Nigeria | SB50 | RV 411 | South Africa | SB81 | RV 361 | South Africa |
| SB20 | RV 329 | South Africa | SB51 | ARC 006 | South Africa | SB82 | ARC 013 | South Africa |
| SB 21 | RV 157 | South Africa | SB52 | 83S-911 | Nigeria | SB83 | ARC 026 | South Africa |
| SB22 | CH 14 | South Africa | SB53 | RV 342 | South Africa | SB84 | TVU 13953 | Nigeria |
| SB23 | TVU 12746 | South Africa | SB54 | ARC 008 | South Africa | SB85 | TVU 13998 | Nigeria |
| SB24 | TVU 9443 | South Africa | SB55 | 90K-284-2 | Nigeria | SB86 | IT00K-1263 | Nigeria |
| SB25 | TVU 12637 | South Africa | SB56 | RV 553 | South Africa | SB87 | RV 555 | South Africa |
| SB26 | TVU 9596 | South Africa | SB57 | RV 213 | South Africa | SB88 | RV 344 | South Africa |
| SB27 | ARC 014 | South Africa | SB58 | GLENDA | South Africa | SB89 | CH 47 | South Africa |
| SB28 | 86D-1010 | Nigeria | SB59 | ARC 005 | South Africa | SB90 | RV 500 | South Africa |
| SB29 | 97K-499-35 | Nigeria | SB60 | RV 320 | South Africa | SB91 | 98K-506-1 | Nigeria |
| SB30 | RV 321 | South Africa | SB61 | RV 502 | South Africa | SB92 | 97K-207-15 | Nigeria |
| SB31 | 99K-494-6 | Nigeria | SB62 | ARC 025 | South Africa |
| Chromosome No. | No. of SNPs | MAF | He | Ho | PIC | FIS |
|---|---|---|---|---|---|---|
| 1 | 518 | 0.7283853 | 0.3462423 | 0.3856441 | 0.3462469 | −0.10820243 |
| 2 | 499 | 0.7300559 | 0.3462424 | 0.3804404 | 0.3428320 | −0.10128670 |
| 3 | 1016 | 0.7357895 | 0.3462425 | 0.3783386 | 0.3433586 | −0.09397598 |
| 4 | 729 | 0.7189496 | 0.3462426 | 0.3922707 | 0.3538314 | −0.10568182 |
| 5 | 654 | 0.7252793 | 0.3462427 | 0.4037193 | 0.3439191 | −0.16628110 |
| 6 | 680 | 0.7239614 | 0.3462428 | 0.4054283 | 0.3518747 | −0.14416821 |
| 7 | 882 | 0.7327958 | 0.3462429 | 0.3667573 | 0.3469722 | −0.05202273 |
| 8 | 630 | 0.7234917 | 0.3462430 | 0.4132967 | 0.3522159 | −0.16792216 |
| 9 | 563 | 0.7329730 | 0.3462431 | 0.3895356 | 0.3357911 | −0.15095750 |
| 10 | 687 | 0.7318918 | 0.3462432 | 0.3770733 | 0.3453611 | −0.08789979 |
| 11 | 639 | 0.7393492 | 0.3462433 | 0.3577612 | 0.3331006 | −0.06902656 |
| Mean | 681.5455 | 0.7293566 | 0.3450427 | 0.3863878 | 0.3450458 | −0.1134023 |
| SE | 46.0903 | 0.001815176 | 0.001949046 | 0.005066684 | 0.001948986 | 0.01172541 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dikane, G.M.H.; Sedibe, M.M. Genetic Heterogeneity in Cowpea Genotypes (Vigna unguiculata L. Walp) Using DArTseq (GBS)-Derived Single Nucleotide Polymorphisms. Genes 2024, 15, 764. https://doi.org/10.3390/genes15060764
Dikane GMH, Sedibe MM. Genetic Heterogeneity in Cowpea Genotypes (Vigna unguiculata L. Walp) Using DArTseq (GBS)-Derived Single Nucleotide Polymorphisms. Genes. 2024; 15(6):764. https://doi.org/10.3390/genes15060764
Chicago/Turabian StyleDikane, Goitsemang Mahlomola Hendry, and Moosa Mahmood Sedibe. 2024. "Genetic Heterogeneity in Cowpea Genotypes (Vigna unguiculata L. Walp) Using DArTseq (GBS)-Derived Single Nucleotide Polymorphisms" Genes 15, no. 6: 764. https://doi.org/10.3390/genes15060764
APA StyleDikane, G. M. H., & Sedibe, M. M. (2024). Genetic Heterogeneity in Cowpea Genotypes (Vigna unguiculata L. Walp) Using DArTseq (GBS)-Derived Single Nucleotide Polymorphisms. Genes, 15(6), 764. https://doi.org/10.3390/genes15060764