
An R2R3-MYB Transcriptional Factor LuMYB314 Associated with the Loss of Petal Pigmentation in Flax (Linum usitatissimum L.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotyping for Petal Color
2.2. Construction of Segregating Pools and Sequencing
2.3. Bulk Segregant Analysis
2.4. Genome-Wide Association Study
2.5. Phylogenetic Analysis
2.6. Gene Cloning and Vector Construction
2.7. Transgenic Analysis
3. Results
3.1. Patterns of Variation in Petal Color
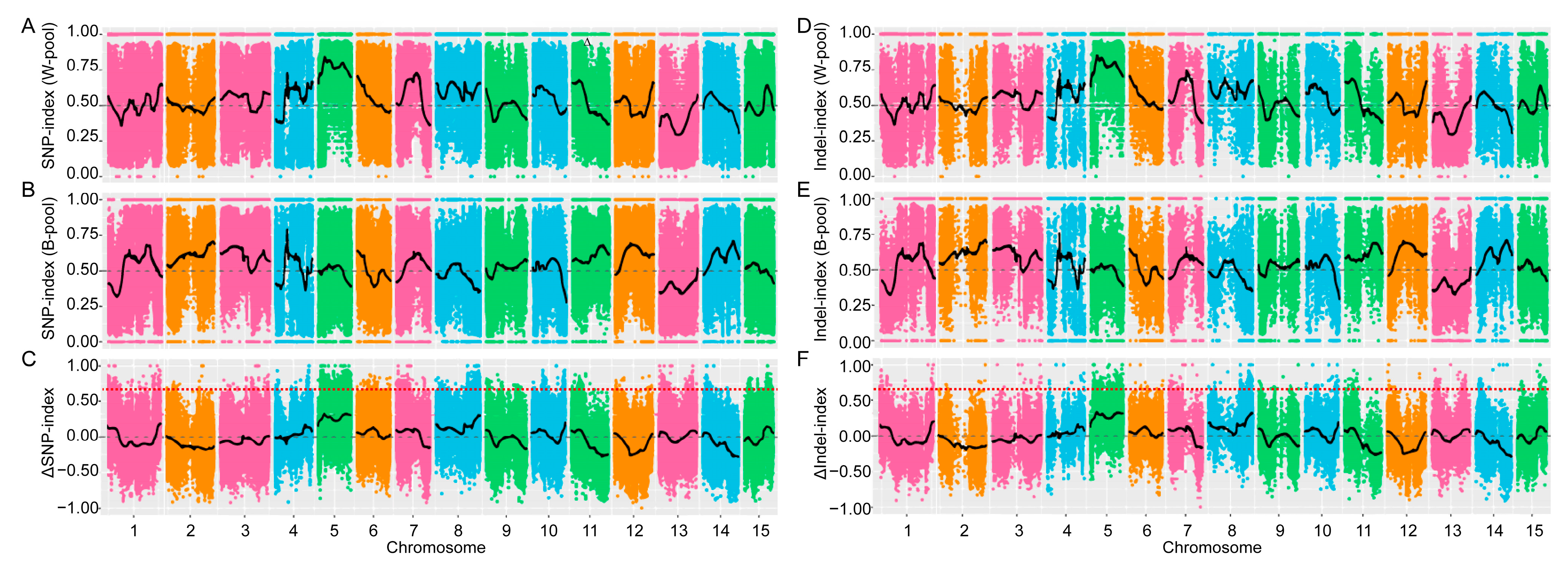
3.2. Whole-Genome Resequencing and Bulk Segregant Analysis
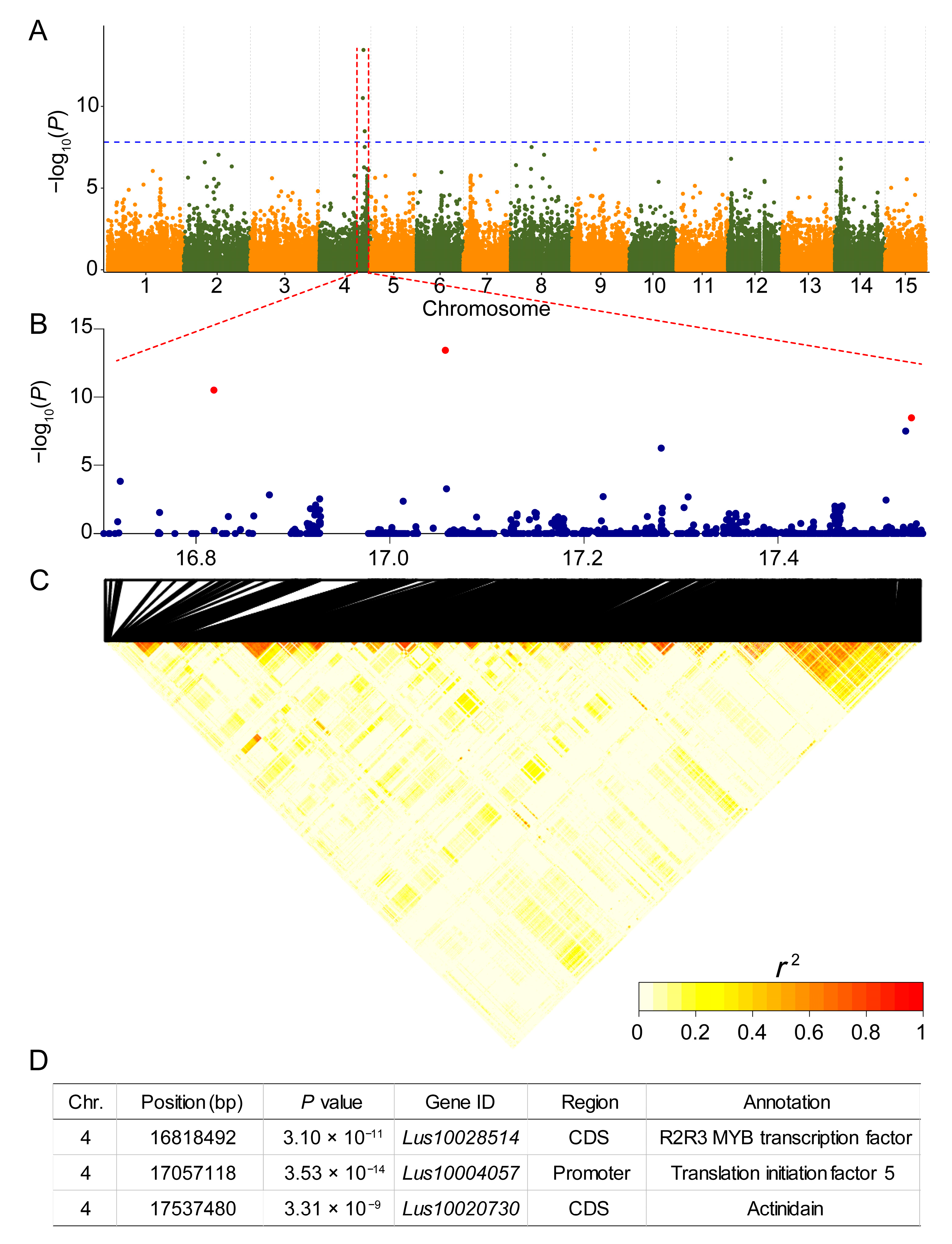
3.3. Identification of a Major QTL for Petal Coloration via a GWAS
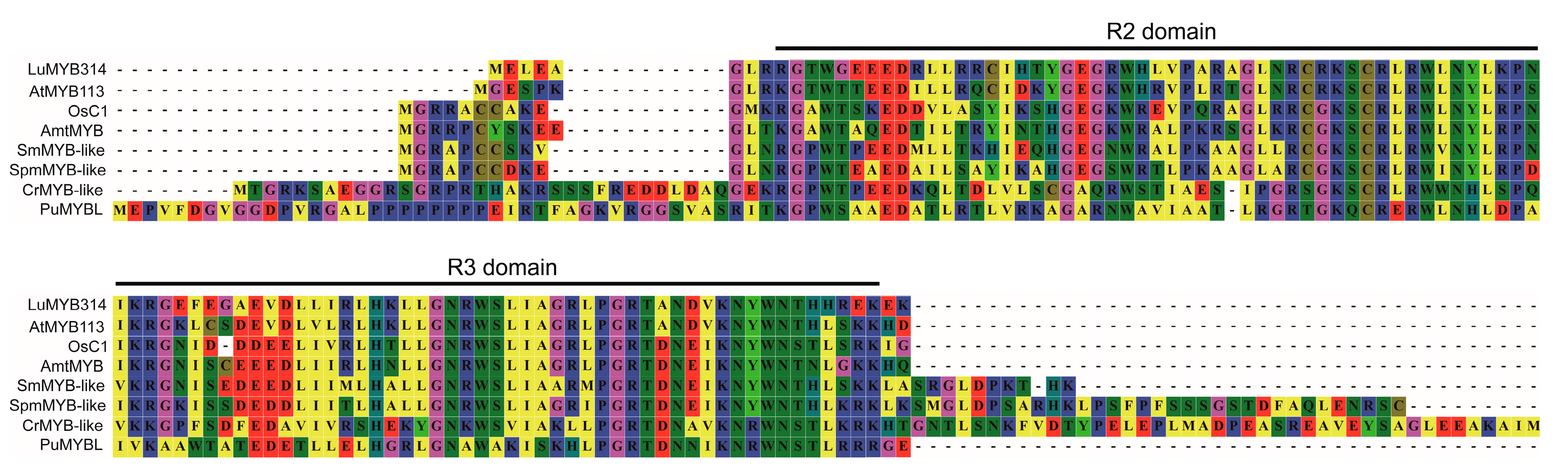
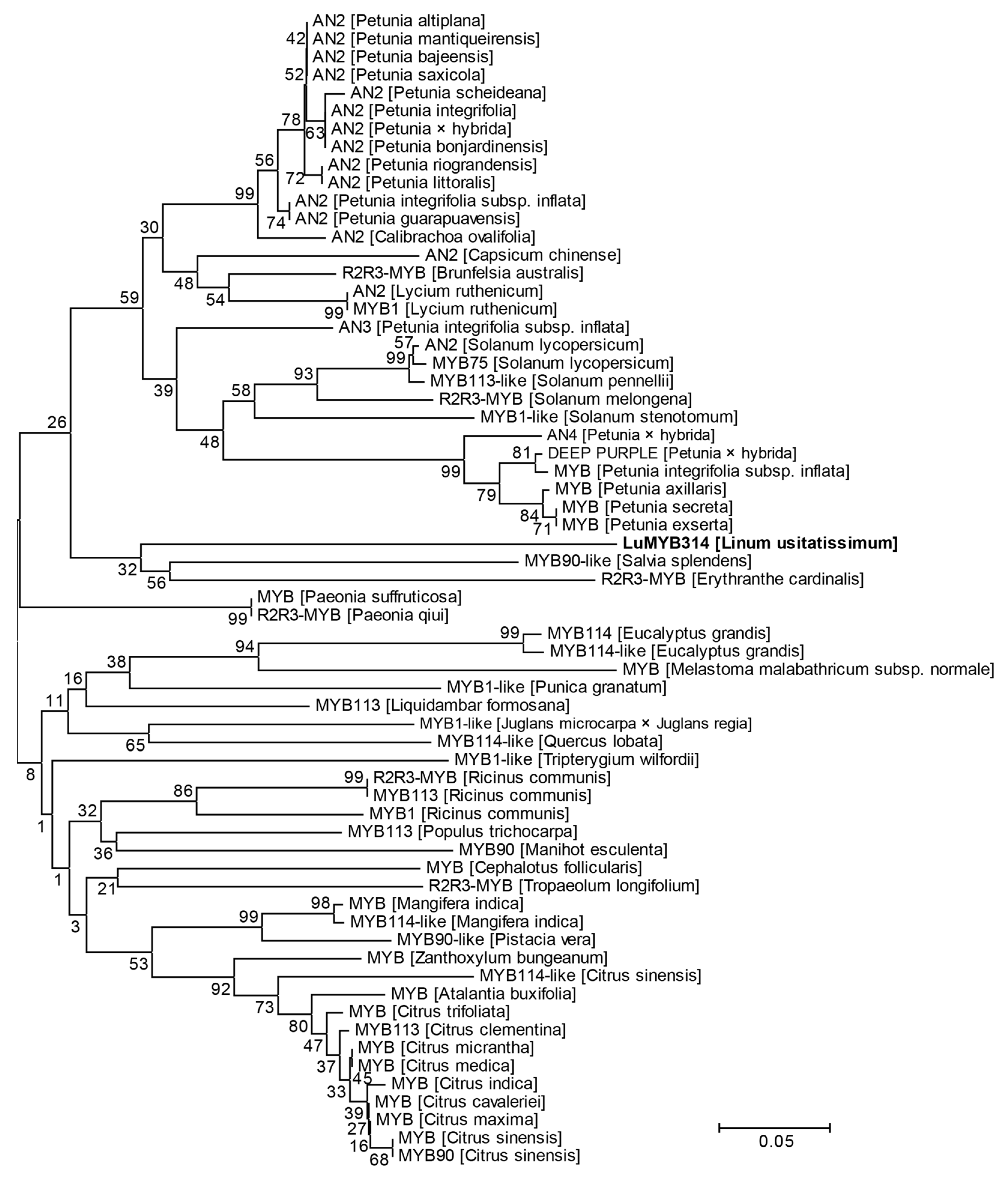
3.4. The Novel LuMYB314 Belongs to a Clade of R2R3 MYB Transcription Factors
3.5. LuMYB314 Associated with Anthocyanin Deposition
4. Discussion
4.1. Natural Variations in Flax Petal Color Were Not Related to Variations in Seed Coat Color
4.2. In Some Weakly Linked Genomic Regions, GWAS Is More Powerful Than BSA-Seq for QTL Detection
4.3. The Subgroup 6 of R2R3-MYB Transcription Factors Is Conserved in Anthocyanin Biosynthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, K.M.; Albert, N.W.; Schwinn, K.E. From landing lights to mimicry: The molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct. Plant Biol. 2012, 39, 619–638. [Google Scholar] [CrossRef] [PubMed]
- Gates, D.J.; Olson, B.J.S.C.; Clemente, T.E.; Smith, S.D. A novel R3 MYB transcriptional repressor associated with the loss of floral pigmentation in Iochroma. New Phytol. 2017, 217, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.A.; Wei, Y.; Meng, Z.; Wang, Y.; Ye, Y.; Wang, Y.; He, H.; Zhou, Q.; Li, Y.; Wang, P.; et al. Increasing floral visitation and hybrid seed production mediated by beauty mark in Gossypium hirsutum. Plant Biotechnol. J. 2022, 20, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, H.D., Jr.; Schemske, D.W. Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers. Nature 2003, 426, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Hoballah, M.E.; Gübitz, T.; Stuurman, J.; Broger, L.; Barone, M.; Mandel, T.; Dell’Olivo, A.; Arnold, M.; Kuhlemeier, C. Single gene-mediated shift in pollinator attraction in Petunia. Plant Cell 2007, 19, 779–790. [Google Scholar] [CrossRef]
- Streisfeld, M.A.; Young, W.N.; Sobel, J.M. Divergent selection drives genetic differentiation in an R2R3-MYB transcription factor that contributes to incipient speciation in Mimulus aurantiacus. PLoS Genet. 2013, 9, e1003385. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, X.; Lin, Z. Convergent loss of anthocyanin pigments is controlled by the same MYB gene in cereals. J. Exp. Bot. 2022, 73, 6089–6102. [Google Scholar] [CrossRef] [PubMed]
- Mol, J.; Jenkins, G.; Schäfer, E.; Weiss, D. Signal perception, transduction, and gene expression involved in anthocyanin biosynthesis. Crit. Rev. Plant Sci. 1996, 15, 525–557. [Google Scholar] [CrossRef]
- Saigo, T.; Wang, T.; Watanabe, M.; Tohge, T. Diversity of anthocyanin and proanthocyanin biosynthesis in land plants. Curr. Opin. Plant Biol. 2020, 55, 93–99. [Google Scholar] [CrossRef]
- Gandikota, M.; Kochko, A.D.; Chen, L.; Ithal, N.; Fauquet, C.; Reddy, A.R. Development of transgenic rice plants expressing maize anthocyanin genes and increased blast resistance. Mol. Breed. 2001, 7, 73–83. [Google Scholar] [CrossRef]
- Gordeeva, E.I.; Shoeva, O.Y.; Khlestkina, E.K. Cold stress response of wheat genotypes having different Rc alleles. Cereal Res. Commun. 2013, 41, 519–526. [Google Scholar] [CrossRef]
- Li, X.; LÜ, X.; Wang, X.; Peng, Q.; Zhang, M.; Ren, M. Biotic and abiotic stress-responsive genes are stimulated to resist drought stress in purple wheat. J. Integr. Agric. 2020, 19, 33–50. [Google Scholar] [CrossRef]
- Shoeva, O.Y.; Gordeeva, E.I.; Arbuzova, V.S.; Khlestkina, E.K. Anthocyanins participate in protection of wheat seedlings from osmotic stress. Cereal Res. Commun. 2017, 45, 47–56. [Google Scholar] [CrossRef]
- Lila, M.A.; Burton-Freeman, B.; Grace, M.; Kalt, W. Unraveling anthocyanin bioavailability for human health. Annu. Rev. Food Sci. Technol. 2016, 7, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Long, T.; Wang, S.; Yang, J. Regulation mechanism of plant pigments biosynthesis: Anthocyanins, carotenoids, and betalains. Metabolites 2022, 12, 871. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, G.G.; Unterlander, N. In through the out door: Biochemical mechanisms affecting flavonoid glycoside catabolism in plants. Plant Sci. 2021, 308, 110904. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Baudry, A.; Heim, M.A.; Dubreucq, B.; Caboche, M.; Weisshaar, B.; Lepiniec, L. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004, 39, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.-M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Paz-Ares, J. MYB transcription factors in plants. Trends Genet. 1997, 13, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Pei, X.; Zhang, H.; Li, X.; Zhang, X.; Zhao, M.; Chiang, V.L.; Sederoff, R.R.; Zhao, X. MYB-mediated regulation of anthocyanin biosynthesis. Int. J. Mol. Sci. 2021, 22, 3103. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jia, K.; Lian, H.; Yang, X.; Li, L.; Yang, H. Jasmonic acid enhancement of anthocyanin accumulation is dependent on phytochrome A signaling pathway under far-red light in Arabidopsis. Biochem. Biophys. Res. Commun. 2014, 454, 78–83. [Google Scholar] [CrossRef] [PubMed]
- LaFountain, A.M.; Yuan, Y. Repressors of anthocyanin biosynthesis. New Phytol. 2021, 231, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Cao, H.; Yuan, S.; Liu, Y.; Lu, J.; Lu, W.; Li, N.; Wang, J.; Zou, J.; Tang, N.; et al. SlMYB75, an MYB-type transcription factor, promotes anthocyanin accumulation and enhances volatile aroma production in tomato fruits. Hortic. Res. 2019, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Chen, N.; Huang, Z.; Li, D.; Zhi, J.; Yu, B.; Liu, X.; Cao, B.; Qiu, Z. Anthocyanin Fruit encodes an R2R3-MYB transcription factor, SlAN2-like, activating the transcription of SlMYBATV to fine-tune anthocyanin content in tomato fruit. New Phytol. 2020, 225, 2048–2063. [Google Scholar] [CrossRef]
- Chen, L.; Hu, B.; Qin, Y.; Hu, G.; Zhao, J. Advance of the negative regulation of anthocyanin biosynthesis by MYB transcription factors. Plant Physiol. Biochem. 2019, 136, 178–187. [Google Scholar] [CrossRef]
- Ma, D.; Constabel, C.P. MYB repressors as regulators of phenylpropanoid metabolism in plants. Trends Plant Sci. 2019, 24, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Sun, M.; Wang, J.; Kawabata, S.; Li, Y. BrMYB4, a suppressor of genes for phenylpropanoid and anthocyanin biosynthesis, is down-regulated by UV-B but not by pigment-inducing sunlight in Turnip cv. Tsuda. Plant Cell Physiol. 2014, 55, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Li, C.; Ma, X.; Luo, K. PtrMYB57 contributes to the negative regulation of anthocyanin and proanthocyanidin biosynthesis in poplar. Plant Cell Rep. 2017, 36, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Qiu, Z.; Wang, X.; Giang, T.V.; Liu, X.; Wang, J.; Wang, X.; Gao, J.; Guo, Y.; Du, Y.; et al. A putative R3 MYB repressor is the candidate gene underlying atroviolacium, a locus for anthocyanin pigmentation in tomato fruit. J. Exp. Bot. 2017, 68, 5745–5758. [Google Scholar] [CrossRef] [PubMed]
- Diederichsen, A. Ex situ collections of cultivated flax (Linum usitatissimum L.) and other species of the genus Linum L. Genet. Resour. Crop Evol. 2007, 54, 661–678. [Google Scholar] [CrossRef]
- Saemundur, S.; Joshua, M.D.; Wong, G.K.S.; Juanjuan, L.; Xia, L.; Deyholos, M.K.; Cronk, Q.C.B. Phylogenetic pinpointing of a paleopolyploidy event within the flax genus (Linum) using transcriptomics. Ann. Bot. 2014, 113, 753–761. [Google Scholar] [CrossRef]
- Soto-Cerda, B.J.; Duguid, S.; Booker, H.; Rowland, G.; Diederichsen, A.; Cloutier, S. Genomic regions underlying agronomic traits in linseed (Linum usitatissimum L.) as revealed by association mapping. J. Integr. Plant Biol. 2014, 56, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Mridula, D.; Rehal, J.; Barnwal, P. Flaxseed: A potential source of food, feed and fiber. Crit. Rev. Food Sci. Nutr. 2011, 51, 210–222. [Google Scholar] [CrossRef]
- Sudarshan, G.P.; Kulkarni, M.; Akhov, L.; Ashe, P.; Shaterian, H.; Cloutier, S.; Rowland, G.; Wei, Y.; Selvaraj, G. QTL mapping and molecular characterization of the classical D locus controlling seed and flower color in Linum usitatissimum (flax). Sci. Rep. 2017, 7, 15751. [Google Scholar] [CrossRef]
- Fulton, T.M.; Chunwongse, J.; Tanksley, S.D. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Report. 1995, 13, 207–209. [Google Scholar] [CrossRef]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Zhu, T.; Luo, M.; Wang, X.; Deyholos, M.K.; et al. Chromosome–scale pseudomolecules refined by optical, physical, and genetic maps in flax. Plant J. 2018, 95, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, Y. Bulk segregation analysis in the NGS era: A review of its teenage years. Plant J. 2022, 109, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhao, T.; You, X.; Jiang, J.; Li, J.; Xu, X. Molecular mapping of the Cf-10 gene by combining SNP/InDel-index and linkage analysis in tomato (Solanum lycopersicum). BMC Plant Biol. 2019, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Luo, W.; Guo, J.; Chen, H.; Akram, W.; Xie, D.; Li, G. Fine mapping and candidate gene analysis of the yellow petal gene ckpc in Chinese kale (Brassica oleracea L. var. alboglabra Bailey) by whole-genome resequencing. Mol. Breed. 2019, 39, 96. [Google Scholar] [CrossRef]
- Guo, D.; Jiang, H.; Yan, W.; Yang, L.; Ye, J.; Wang, Y.; Yan, Q.; Chen, J.; Gao, Y.; Duan, L.; et al. Resequencing 200 flax cultivated accessions identifies candidate genes related to seed size and weight and reveals signatures of artificial selection. Front. Plant Sci. 2020, 10, 1682. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef]
- Shin, J.H.; Blay, S.; Mcneney, B.; Graham, J. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 2006, 16, 1–9. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Maleki, S.S.; Mohammadi, K.; Ji, K. Influencing factors of genetic transformation efficiency in tobacco (Nicotiana tabacum ‘NC89’) using Agrobacterium tumefacience. J. Nanjing For. Univ. 2018, 42, 27–34. [Google Scholar] [CrossRef]
- Zhang, W.; Ning, G.; Lv, H.; Liao, L.; Bao, M. Single MYB-type transcription factor AtCAPRICE: A new efficient tool to engineer the production of anthocyanin in tobacco. Biochem. Biophys. Res. Commun. 2009, 388, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Sagawa, J.M.; Frost, L.; Vela, J.P.; Bradshaw, H.D., Jr. Transcriptional control of floral anthocyanin pigmentation in monkeyflowers (Mimulus). New Phytol. 2015, 204, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Li, J.; Zhang, X.; Dai, Z.; Zhou, W.; Su, J.; Sun, J. Systematic analysis of MYB transcription factors and the role of LuMYB216 in regulating anthocyanin biosynthesis in the flowers of flax (Linum usitatissimum L.). J. Integr. Agric. 2023, 22, 2335–2345. [Google Scholar] [CrossRef]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Hyten, D.L.; Choi, I.-Y.; Song, Q.; Shoemaker, R.C.; Nelson, R.L.; Costa, J.M.; Specht, J.E.; Cregan, P.B. Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics 2007, 175, 1937–1944. [Google Scholar] [CrossRef]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-wide association study of 12 agronomic traits in peach. Nat. Commun. 2016, 7, 13246. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Lian, Q.; Zhang, Z.; Fu, Q.; He, Y.; Ma, S.; Ruggieri, V.; Monforte, A.J.; Wang, P.; Julca, I.; et al. A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat. Genet. 2019, 51, 1607–1615. [Google Scholar] [CrossRef]
- Guo, S.; Zhao, S.; Sun, H.; Wang, X.; Wu, S.; Lin, T.; Ren, Y.; Gao, L.; Deng, Y.; Zhang, J.; et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 2019, 51, 1616–1623. [Google Scholar] [CrossRef]
- Liu, D.; Yang, H.; Yuan, Y.; Zhu, H.; Zhang, M.; Wei, X.; Sun, D.; Wang, X.; Yang, S.; Yang, L. Comparative transcriptome analysis provides insights into yellow rind formation and preliminary mapping of the Clyr (yellow rind) gene in watermelon. Front. Plant Sci. 2020, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Yun, W.; Jing, W.; Yiping, X.; Liangsheng, Z.; Hai, D. Evolution and functional diversification of R2R3-MYB transcription factors in plants. Hortic. Res. 2022, 9, uhac058. [Google Scholar] [CrossRef]
- Du, H.; Liang, Z.; Zhao, S.; Nan, M.; Tran, L.-S.P.; Lu, K.; Huang, Y.; Li, J. The evolutionary history of R2R3-MYB proteins across 50 eukaryotes: New insights into subfamily classification and expansion. Sci. Rep. 2015, 5, 11037. [Google Scholar] [CrossRef]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S.; Ishizaki, K.; Yamaoka, S.; Nishihama, R.; Nakamura, Y.; Berger, F.; et al. Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 2017, 171, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Kranz, H.D.; Denekamp, M.; Greco, R.; Jin, H.; Leyva, A.; Meissner, R.C.; Petroni, K.; Urzainqui, A.; Bevan, M.; Martin, C.; et al. Towards functional characterisation of the members of the R2R3-MYB gene family from Arabidopsis thaliana. Plant J. 1998, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Q.; Feng, K.; Yu, X.; Xiong, A. DcMYB113, a root-specific R2R3-MYB, conditions anthocyanin biosynthesis and modification in carrot. Plant Biotechnol. J. 2020, 18, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Zhou, F.; Li, W.; Yu, Y.; Guan, Z.; Zhang, B.; Guo, W. The R2R3-MYB transcription factor GaPC controls petal coloration in cotton. Crop J. 2023, 11, 1319–1330. [Google Scholar] [CrossRef]
- Li, J.; Jiang, S.; Yang, G.; Xu, Y.; Li, L.; Yang, F. RNA-sequencing analysis reveals novel genes involved in the different peel color formation in eggplant. Hortic. Res. 2023, 10, uhad181. [Google Scholar] [CrossRef]
- Yang, G.; Li, L.; Wei, M.; Li, J.; Yang, F. SmMYB113 Is a key transcription factor responsible for compositional variation of anthocyanin and color diversity among eggplant peels. Front. Plant Sci. 2022, 13, 843996. [Google Scholar] [CrossRef]
- Jiang, C.; Rao, G. Insights into the diversification and evolution of R2R3-MYB transcription factors in plants. Plant Physiol. 2020, 183, 637–655. [Google Scholar] [CrossRef]
- Chang, X.; Xie, S.; Wei, L.; Lu, Z.; Chen, Z.; Chen, F.; Lai, Z.; Lin, Z.; Zhang, L. Origins and stepwise expansion of R2R3-MYB transcription factors for the terrestrial adaptation of plants. Front. Plant Sci. 2020, 11, 575360. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, D.; Jiang, H.; Xie, L. An R2R3-MYB Transcriptional Factor LuMYB314 Associated with the Loss of Petal Pigmentation in Flax (Linum usitatissimum L.). Genes 2024, 15, 511. https://doi.org/10.3390/genes15040511
Guo D, Jiang H, Xie L. An R2R3-MYB Transcriptional Factor LuMYB314 Associated with the Loss of Petal Pigmentation in Flax (Linum usitatissimum L.). Genes. 2024; 15(4):511. https://doi.org/10.3390/genes15040511
Chicago/Turabian StyleGuo, Dongliang, Haixia Jiang, and Liqiong Xie. 2024. "An R2R3-MYB Transcriptional Factor LuMYB314 Associated with the Loss of Petal Pigmentation in Flax (Linum usitatissimum L.)" Genes 15, no. 4: 511. https://doi.org/10.3390/genes15040511
APA StyleGuo, D., Jiang, H., & Xie, L. (2024). An R2R3-MYB Transcriptional Factor LuMYB314 Associated with the Loss of Petal Pigmentation in Flax (Linum usitatissimum L.). Genes, 15(4), 511. https://doi.org/10.3390/genes15040511