
Biogeographical Ancestry Analyses Using the ForenSeqTM DNA Signature Prep Kit and Multiple Prediction Tools

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Library Preparation and Sequencing
2.3. Analysis of the Sequence Data
2.4. Population Structure of the Norwegian Reference Population
2.5. Biogeographical Ancestry Prediction
2.6. UAS
2.7. FROG-kb
2.8. GenoGeographer
3. Results
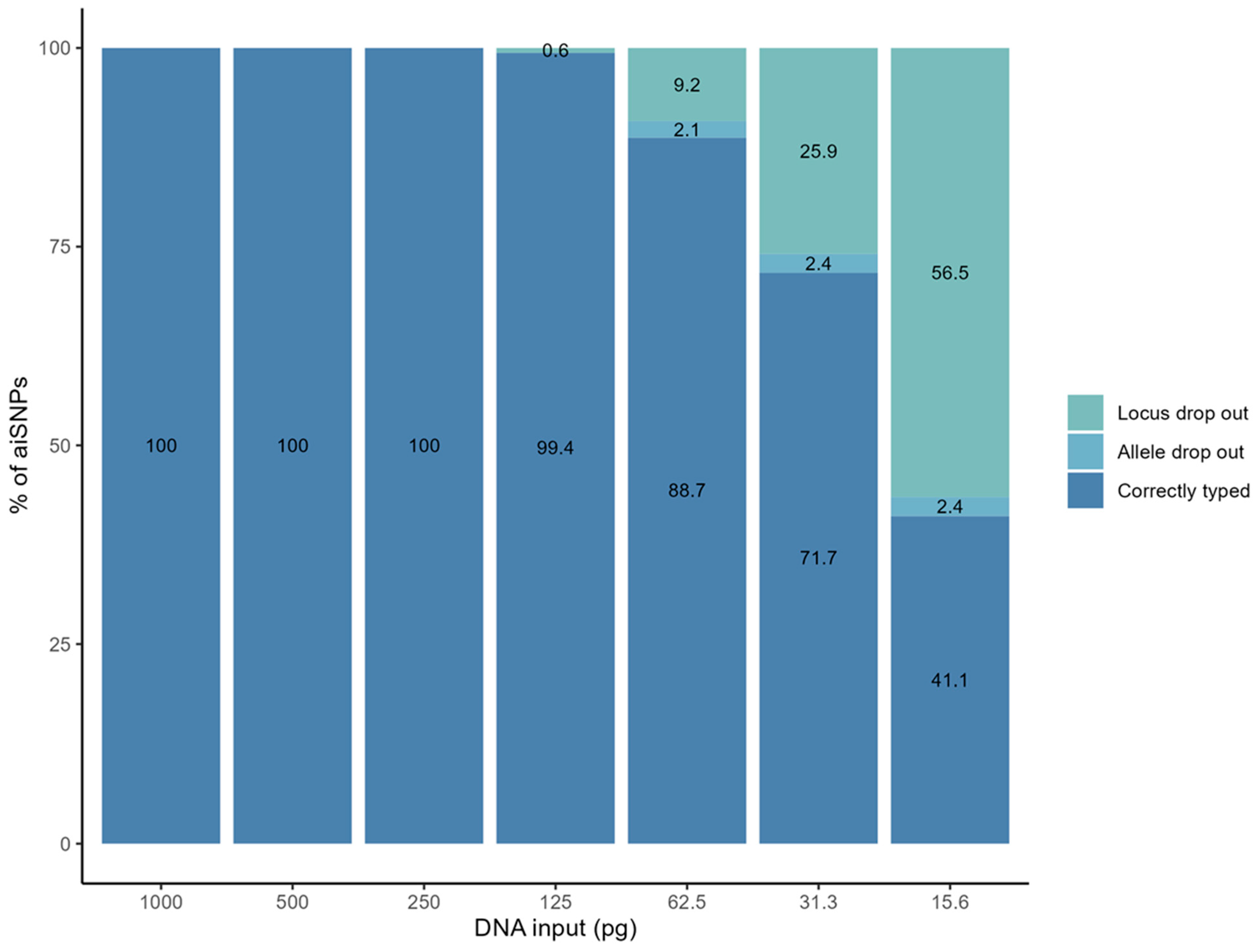
3.1. Genotyping Performance of the aiSNPs Using the ForenSeq™ DNA Signature Prep Kit
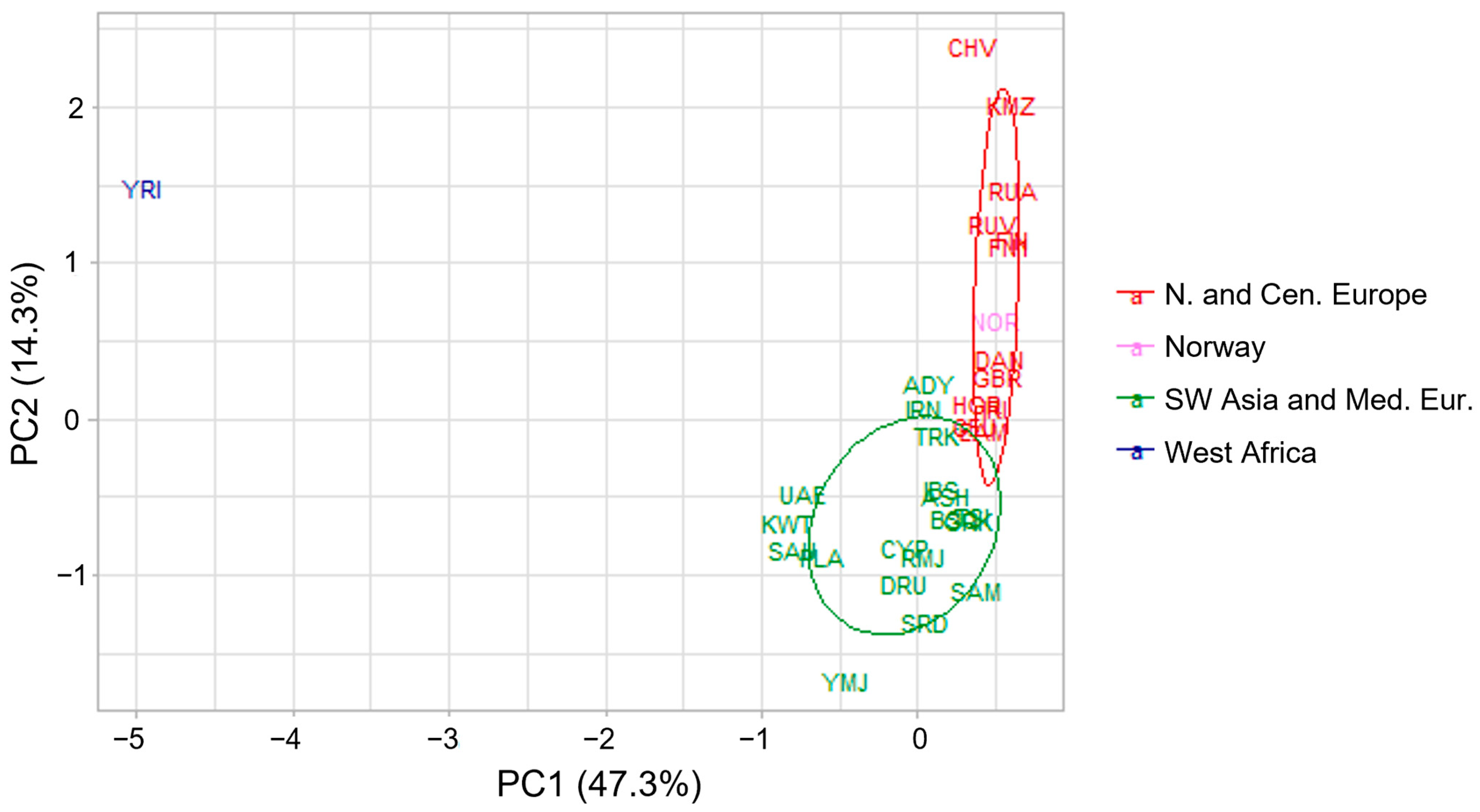
3.2. Genetic Structure of the Norwegian Reference Population
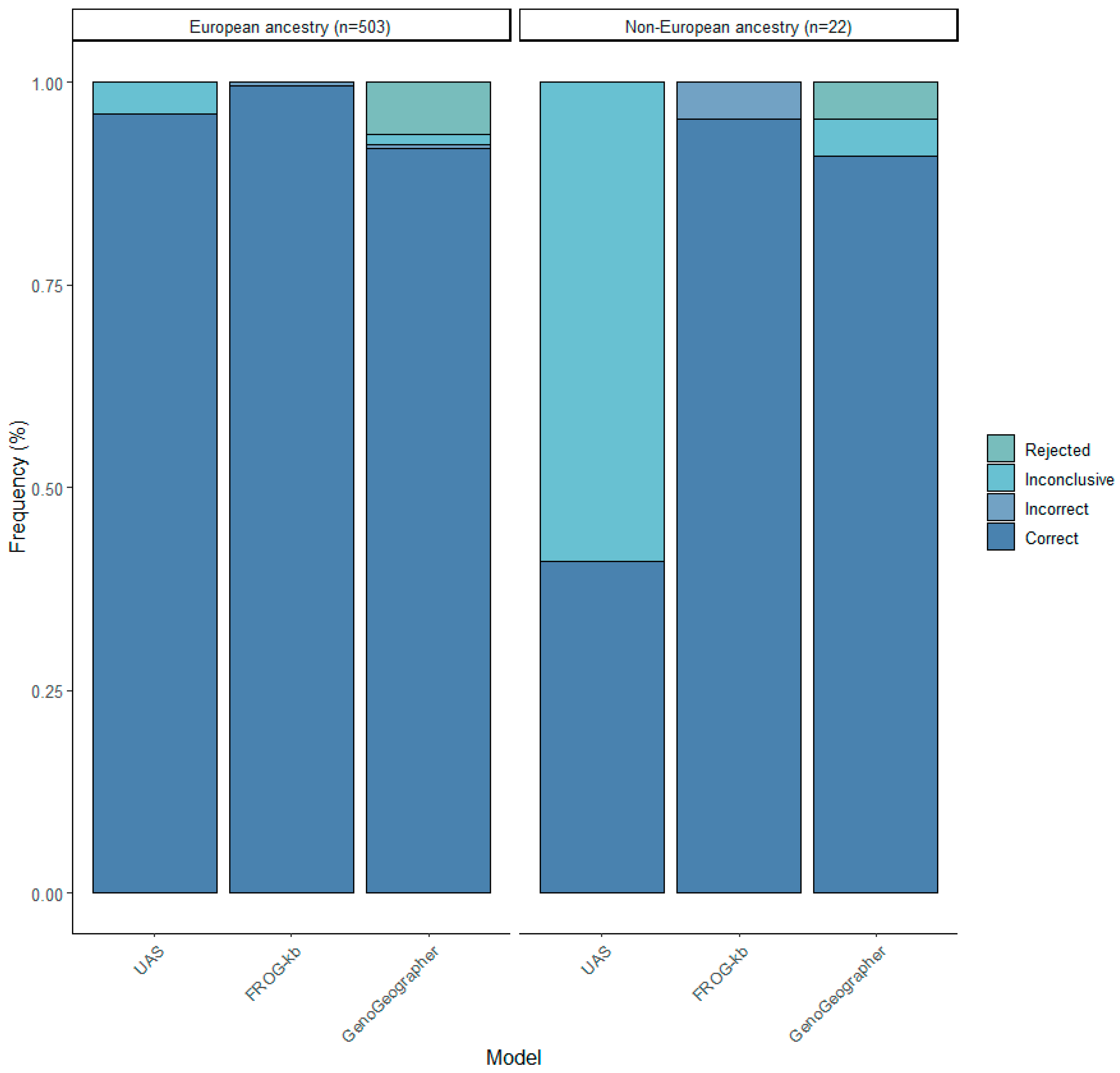
3.3. Biogeographical Ancestry Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phillips, C. Forensic genetic analysis of bio-geographical ancestry. Forensic Sci. Int. Genet. 2015, 18, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Kayser, M. Forensic DNA Phenotyping: Predicting human appearance from crime scene material for investigative purposes. Forensic Sci. Int. Genet. 2015, 18, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.M.; Prainsack, B.; Kayser, M. The use of forensic DNA phenotyping in predicting appearance and biogeographic ancestry. Dtsch. Arztebl. Int. 2019, 116, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; McNevin, D.; Kidd, K.K.; Lagacé, R.; Wootton, S.; de la Puente, M.; Freire-Aradas, A.; Mosquera-Miguel, A.; Eduardoff, M.; Gross, T.; et al. MAPlex—A massively parallel sequencing ancestry analysis multiplex for Asia-Pacific populations. Forensic Sci. Int. Genet. 2019, 42, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Podini, D.S.; Lagacé, R.; Chang, J.; Wootton, S.; Haigh, E.; Soundararajan, U. Evaluating 130 microhaplotypes across a global set of 83 populations. Forensic Sci. Int. Genet. 2017, 29, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Phillips, C.; Pinto, N.; Santos, C.; Sebd, S. Straightforward Inference of Ancestry and Admixture Proportions through Ancestry-Informative Insertion Deletion Multiplexing. PLoS ONE 2012, 7, 29684. [Google Scholar] [CrossRef] [PubMed]
- Børsting, C.; Morling, N. Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 2015, 18, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; De La Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.E.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq basic tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Madrid, L.; Xavier, C.; De La Puente, M.; Hohoff, C.; Phillips, C.; Kayser, M.; Parson, W. Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes 2020, 11, 708. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.C.; Alvarez, M.L.; Davis, C.P.; Guzmán, E.; Han, Y.; Way, L.; Walichiewicz, P.; Silva, D.; Pham, N.; Caves, G.; et al. Developmental validation of the MiSeq FGx Forensic Genomics System for Targeted Next Generation Sequencing in Forensic DNA Casework and Database Laboratories. Forensic Sci. Int. Genet. 2017, 28, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Yu, J.; Zhang, L.; Li, J. Massively parallel sequencing of forensic STRs and SNPs using the Illumina® ForenSeqTM DNA Signature Prep Kit on the MiSeq FGxTM Forensic Genomics System. Forensic Sci. Int. Genet. 2017, 31, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Furtado, M.R.; Fang, R.; Madbouly, A.; Maiers, M.; Middha, M.; Friedlaender, F.R.; Kidd, J.R. Progress toward an efficient panel of SNPs for ancestry inference. Forensic Sci. Int. Genet. 2014, 10, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Pakstis, A.J.; Kang, L.; Liu, L.; Zhang, Z.; Jin, T.; Grigorenko, E.L.; Wendt, F.R.; Budowle, B.; Hadi, S.; Al Qahtani, M.S.; et al. Increasing the reference populations for the 55 AISNP panel: The need and benefits. Int. J. Legal Med. 2017, 131, 913–917. [Google Scholar] [CrossRef]
- Pakstis, A.J.; Gurkan, C.; Dogan, M.; Balkaya, H.E.; Dogan, S.; Neophytou, P.I.; Cherni, L.; Boussetta, S.; Khodjet-El-Khil, H.; ElGaaied, A.B.A.; et al. Genetic relationships of European, Mediterranean, and SW Asian populations using a panel of 55 AISNPs. Eur. J. Hum. Genet. 2019, 27, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Rajeevan, H.; Soundararajan, U.; Pakstis, A.J.; Kidd, K.K. Introducing the Forensic Research/Reference on Genetics knowledge base, FROG-kb. Investig. Genet. 2012, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Rajeevan, H.; Soundararajan, U.; Pakstis, A.J.; Kidd, K.K. FrogAncestryCalc: A standalone batch likelihood computation tool for ancestry inference panels catalogued in FROG-kb. Forensic Sci. Int. Genet. 2020, 46, 102237. [Google Scholar] [CrossRef] [PubMed]
- Tvedebrink, T.; Eriksen, P.S.; Mogensen, H.S.; Morling, N. GenoGeographer—A tool for genogeographic inference. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e463–e465. [Google Scholar] [CrossRef]
- Frégeau, C.J. A multiple predictive tool approach for phenotypic and biogeographical ancestry inferences. Can. Soc. Forensic Sci. J. 2022, 55, 71–99. [Google Scholar] [CrossRef]
- Resutik, P.; Aeschbacher, S.; Krützen, M.; Kratzer, A.; Haas, C.; Phillips, C.; Arora, N. Comparative evaluation of the MAPlex, Precision ID Ancestry Panel, and VISAGE Basic Tool for biogeographical ancestry inference. Forensic Sci. Int. Genet. 2023, 64, 102850. [Google Scholar] [CrossRef] [PubMed]
- Salvo, N.M.; Janssen, K.; Kirsebom, M.K.; Meyer, O.S.; Berg, T.; Olsen, G.H. Predicting eye and hair colour in a Norwegian population using Verogen’s ForenSeqTM DNA signature prep kit. Forensic Sci. Int. Genet. 2022, 56, 102620. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, H.S.; Tvedebrink, T.; Børsting, C.; Pereira, V.; Morling, N. Ancestry prediction efficiency of the software GenoGeographer using a z-score method and the ancestry informative markers in the Precision ID Ancestry Panel. Forensic Sci. Int. Genet. 2020, 44, 102154. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Churchill, J.D.; Novroski, N.M.M.; King, J.L.; Seah, L.H.; Budowle, B. Population and performance analyses of four major populations with Illumina’s FGx Forensic Genomics System. Forensic Sci. Int. Genet. 2017, 30, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Sidstedt, M.; Junker, K.; Forsberg, C.; Boiso, L.; Rådström, P.; Ansell, R.; Hedman, J. In-house validation of MPS-based methods in a forensic laboratory. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 635–636. [Google Scholar] [CrossRef]
- Frégeau, C.J. Validation of the Verogen ForenSeqTM DNA Signature Prep kit/Primer Mix B for phenotypic and biogeographical ancestry predictions using the Micro MiSeq® Flow Cells. Forensic Sci. Int. Genet. 2021, 53, 102533. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Jani, K.; Khosla, P.; Butler, E.; Siegel, D.; Wurmbach, E. Evaluation of ForenSeqTM Signature Prep Kit B on predicting eye and hair coloration as well as biogeographical ancestry by using Universal Analysis Software (UAS) and available web-tools. Electrophoresis 2019, 40, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Tvedebrink, T.; Eriksen, P.S.; Mogensen, H.S.; Morling, N. Weight of the evidence of genetic investigations of ancestry informative markers. Theor. Popul. Biol. 2018, 120, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.Y.Y.; Gahan, M.E.; McNevin, D. Prediction of biogeographical ancestry in admixed individuals. Forensic Sci. Int. Genet. 2018, 36, 104–111. [Google Scholar] [CrossRef] [PubMed]
- García, O.; Ajuriagerra, J.A.; Alday, A.; Alonso, S.; Pérez, J.A.; Soto, A.; Uriarte, I.; Yurrebaso, I. Frequencies of the precision ID ancestry panel markers in Basques using the Ion Torrent PGMTM platform. Forensic Sci. Int. Genet. 2017, 31, e1–e4. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvo, N.M.; Olsen, G.-H.; Berg, T.; Janssen, K. Biogeographical Ancestry Analyses Using the ForenSeqTM DNA Signature Prep Kit and Multiple Prediction Tools. Genes 2024, 15, 510. https://doi.org/10.3390/genes15040510
Salvo NM, Olsen G-H, Berg T, Janssen K. Biogeographical Ancestry Analyses Using the ForenSeqTM DNA Signature Prep Kit and Multiple Prediction Tools. Genes. 2024; 15(4):510. https://doi.org/10.3390/genes15040510
Chicago/Turabian StyleSalvo, Nina Mjølsnes, Gunn-Hege Olsen, Thomas Berg, and Kirstin Janssen. 2024. "Biogeographical Ancestry Analyses Using the ForenSeqTM DNA Signature Prep Kit and Multiple Prediction Tools" Genes 15, no. 4: 510. https://doi.org/10.3390/genes15040510
APA StyleSalvo, N. M., Olsen, G.-H., Berg, T., & Janssen, K. (2024). Biogeographical Ancestry Analyses Using the ForenSeqTM DNA Signature Prep Kit and Multiple Prediction Tools. Genes, 15(4), 510. https://doi.org/10.3390/genes15040510