

Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing


Abstract
1. Introduction
2. Methods
2.1. Overview, Study Design, and Ethics
2.2. Subjects and Case Classifications
2.3. Genetic Testing Practices and Defining Genetic Diagnoses
2.4. Additional Data
2.5. Statistical Analyses and Screening Performance
2.6. Decision Curve Analysis
3. Results
3.1. Cohort Description
3.2. Genetic Testing and Genetic Diagnostic Outcomes
3.3. ECA Status and Genetic Diagnoses Identified
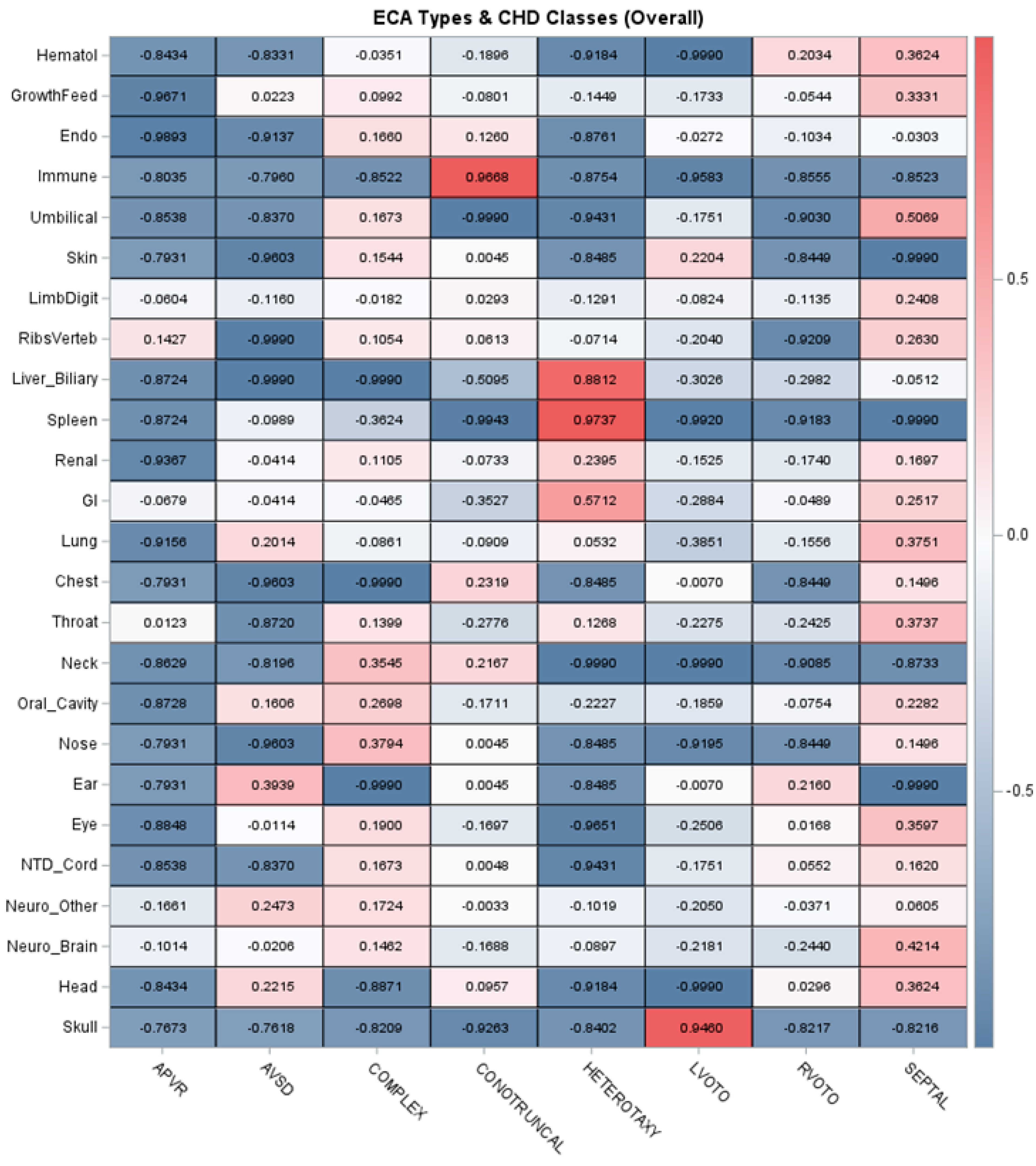
3.4. ECA Patterns and Associations with Genetic Diagnoses Identified
3.5. Screening Performance of ECA Status
3.6. Decision Curve Analysis
3.7. Types of Genetic Diagnoses and Informing Future Genetic Testing Strategies
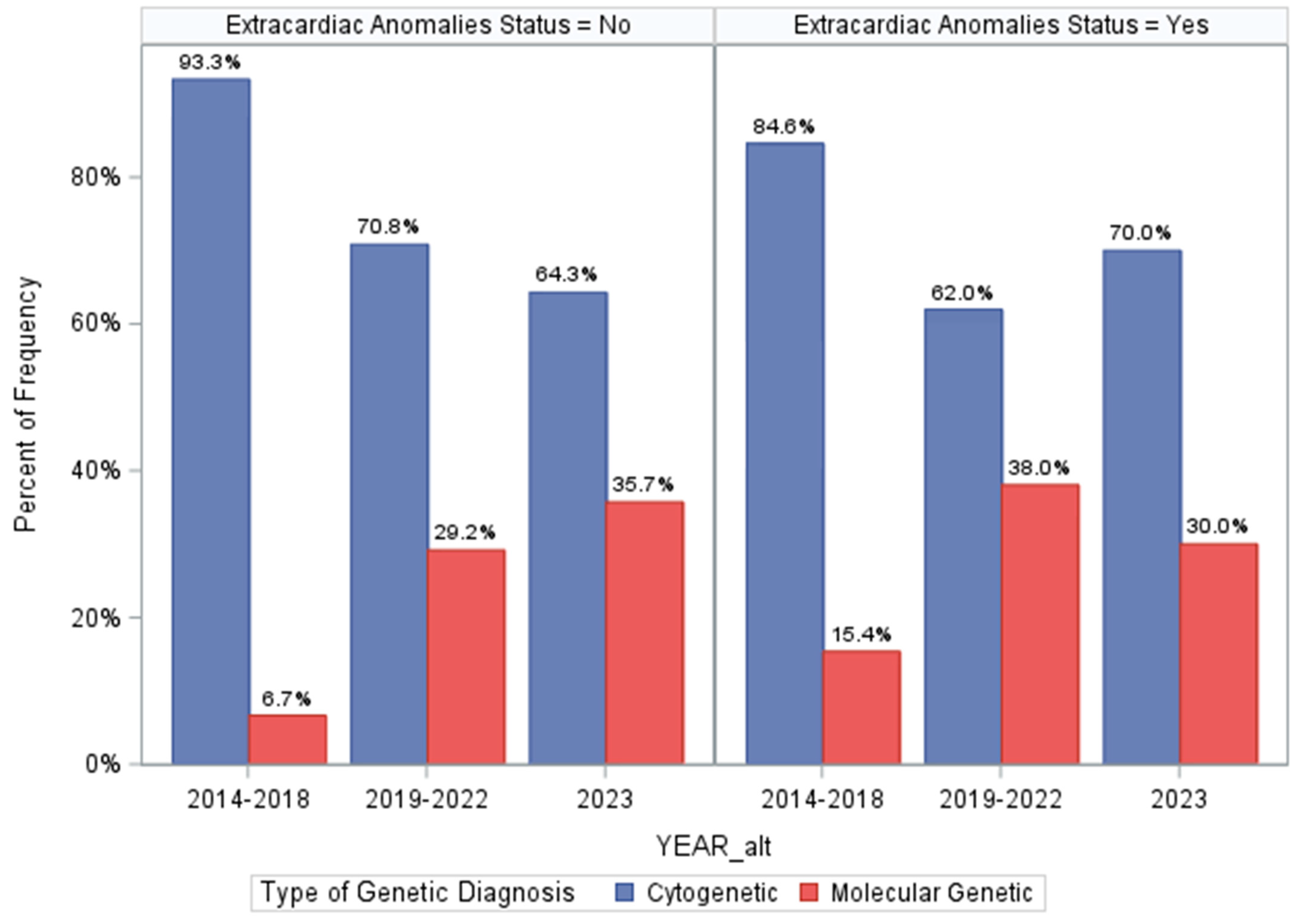
3.8. Assessing Differences in Genetic Diagnosis over Program Time Periods
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cowan, J.R.; Ware, S.M. Genetics and genetic testing in congenital heart disease. Clin. Perinatol. 2015, 42, 373–393. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef] [PubMed]
- Durbin, M.D.; Helvaty, L.R.; Li, M.; Border, W.; Fitzgerald-Butt, S.; Garg, V.; Geddes, G.C.; Helm, B.M.; Lalani, S.R.; McBride, K.L.; et al. A multicenter cross-sectional study in infants with congenital heart defects demonstrates high diagnostic yield of genetic testing but variable evaluation practices. Genet. Med. Open 2023, 1, 100814. [Google Scholar] [CrossRef]
- Ison, H.E.; Griffin, E.L.; Parrott, A.; Shikany, A.R.; Meyers, L.; Thomas, M.J.; Syverson, E.; Demo, E.M.; Fitzgerald, K.K.; Fitzgerald-Butt, S.; et al. Genetic counseling for congenital heart disease—Practice resource of the National Society of Genetic Counselors. J. Genet. Couns. 2022, 31, 9–33. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Muiño Mosquera, L. Genetic Testing in Patients with Congenital Heart Disease: You Do No Harm When Using the Right Tools! Circ. Genom. Precis. Med. 2023, 16, e004104. [Google Scholar] [CrossRef] [PubMed]
- Durbin, M.D.; Fairman, K.; Helvaty, L.R.; Huang, M.; Li, M.; Abreu, D.; Geddes, G.C.; Helm, B.M.; Landis, B.J.; McEntire, A.; et al. Genetic Testing Guidelines Impact Care in Newborns with Congenital Heart Defects. J. Pediatr. 2023, 260, 113495. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.A.; Hinton, R.B.; Miller, E.M.; Sund, K.L.; Ruschman, J.G.; Ware, S.M. Genetic testing practices in infants with congenital heart disease. Congenit. Heart Dis. 2014, 9, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Fakhro, K.A.; Choi, M.; Ware, S.M.; Belmont, J.W.; Towbin, J.A.; Lifton, R.P.; Khokha, M.K.; Brueckner, M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. USA 2011, 108, 2915–2920. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; McBride, K.L.; Bleyl, S.B.; Bowles, N.E.; Border, W.L.; Garg, V.; Smolarek, T.A.; Lalani, S.R.; Ware, S.M. Rationale for the Cytogenomics of Cardiovascular Malformations Consortium: A Phenotype Intensive Registry Based Approach. J. Cardiovasc. Dev. Dis. 2015, 2, 76–92. [Google Scholar] [CrossRef]
- Lalani, S.R.; Shaw, C.; Wang, X.; Patel, A.; Patterson, L.W.; Kolodziejska, K.; Szafranski, P.; Ou, Z.; Tian, Q.; Kang, S.H.; et al. Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Eur. J. Hum. Genet. 2013, 21, 173–181. [Google Scholar] [CrossRef]
- Lander, J.; Ware, S.M. Copy number variation in congenital heart defects. Curr. Genet. Med. Rep. 2014, 2, 168–178. [Google Scholar] [CrossRef]
- Landis, B.J.; Ware, S.M. The Current Landscape of Genetic Testing in Cardiovascular Malformations: Opportunities and Challenges. Front. Cardiovasc. Med. 2016, 3, 22. [Google Scholar] [CrossRef]
- Ware, S.M.; Jefferies, J.L. New Genetic Insights into Congenital Heart Disease. J. Clin. Exp. Cardiolog. 2012, S8, 003. [Google Scholar]
- Helm, B.M.; Freeze, S.L. Genetic Evaluation and Use of Chromosome Microarray in Patients with Isolated Heart Defects: Benefits and Challenges of a New Model in Cardiovascular Care. Front. Cardiovasc. Med. 2016, 3, 19. [Google Scholar] [CrossRef]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Helm, B.M.; Landis, B.J.; Ware, S.M. Genetic Evaluation of Inpatient Neonatal and Infantile Congenital Heart Defects: New Findings and Review of the Literature. Genes 2021, 12, 1244. [Google Scholar] [CrossRef]
- Shikany, A.R.; Landis, B.J.; Parrott, A.; Miller, E.M.; Coyan, A.; Walters, L.; Hinton, R.B.; Goldenberg, P.; Ware, S.M. A Comprehensive Clinical Genetics Approach to Critical Congenital Heart Disease in Infancy. J. Pediatr. 2020, 227, 231–238.e14. [Google Scholar] [CrossRef]
- Geddes, G.C.; Basel, D.; Frommelt, P.; Kinney, A.; Earing, M. Genetic Testing Protocol Reduces Costs and Increases Rate of Genetic Diagnosis in Infants with Congenital Heart Disease. Pediatr. Cardiol. 2017, 38, 1465–1470. [Google Scholar] [CrossRef]
- Landis, B.J.; Helvaty, L.R.; Geddes, G.C.; Lin, J.I.; Yatsenko, S.A.; Lo, C.W.; Border, W.L.; Wechsler, S.B.; Murali, C.N.; Azamian, M.S.; et al. A Multicenter Analysis of Abnormal Chromosomal Microarray Findings in Congenital Heart Disease. J. Am. Heart Assoc. 2023, 12, e029340. [Google Scholar] [CrossRef]
- Landis, B.J.; Helm, B.M.; Herrmann, J.L.; Hoover, M.C.; Durbin, M.D.; Elmore, L.R.; Huang, M.; Johansen, M.; Li, M.; Przybylowski, L.F.; et al. Learning to Crawl: Determining the Role of Genetic Abnormalities on Postoperative Outcomes in Congenital Heart Disease. J. Am. Heart Assoc. 2022, 11, e026369. [Google Scholar] [CrossRef]
- McAfee, K.; Rosenow, W.T.; Cherny, S.; Collins, C.A.; Balmert, L.C.; Webster, G. Abnormal Microarray, Clinical Outcomes, and Surgical Risk Scores in Young Children with Cardiac Disease. Pediatr. Cardiol. 2021, 42, 1785–1791. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Kim, J.J.; Gelb, B.D.; Helm, B.M.; Kannankeril, P.J.; Semsarian, C.; Sturm, A.C.; Tristani-Firouzi, M.; Ware, S.M. Genetic Testing for Heritable Cardiovascular Diseases in Pediatric Patients: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2021, 14, e000086. [Google Scholar] [CrossRef]
- De Backer, J.; Bondue, A.; Budts, W.; Evangelista, A.; Gallego, P.; Jondeau, G.; Loeys, B.; Peña, M.L.; Teixido-Tura, G.; van de Laar, I.; et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human Genetics. Eur. J. Prev. Cardiol. 2020, 27, 1423–1435. [Google Scholar]
- Chaix, M.A.; Andelfinger, G.; Khairy, P. Genetic testing in congenital heart disease: A clinical approach. World J. Cardiol. 2016, 8, 180–191. [Google Scholar] [CrossRef]
- Egbe, A.; Lee, S.; Ho, D.; Uppu, S.; Srivastava, S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann. Pediatr. Cardiol. 2014, 7, 86–91. [Google Scholar] [CrossRef]
- Botto, L.D.; Lin, A.E.; Riehle-Colarusso, T.; Malik, S.; Correa, A. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 714–727. [Google Scholar] [CrossRef]
- Jones, K.L.; Adam, M.P. Evaluation and diagnosis of the dysmorphic infant. Clin. Perinatol. 2015, 42, 243–261, vii–viii. [Google Scholar] [CrossRef]
- Adam, M.; Hudgins, L. The Importance of Minor Anomalies in the Evaluation of the Newborn. NeoReviews 2003, 4, e99–e104. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Linn, S.; Grunau, P.D. New patient-oriented summary measure of net total gain in certainty for dichotomous diagnostic tests. Epidemiol. Perspect. Innov. 2006, 3, 11. [Google Scholar] [CrossRef]
- Mitchell, A.J. Sensitivity × PPV is a recognized test called the clinical utility index (CUI+). Eur. J. Epidemiol. 2011, 26, 251–252. [Google Scholar] [CrossRef]
- Larner, A.J. New unitary metrics for dementia test accuracy studies. Prog. Neurol. Psychiatry 2019, 23, 21–25. [Google Scholar] [CrossRef]
- Djulbegovic, B.; Hozo, I. Using Decision Curve Analysis to Evaluate Testing and/or Predictive Modeling. Cancer Treat. Res. 2023, 189, 77–84. [Google Scholar]
- Fitzgerald, M.; Saville, B.R.; Lewis, R.J. Decision curve analysis. JAMA 2015, 313, 409–410. [Google Scholar] [CrossRef]
- Van Calster, B.; Wynants, L.; Verbeek, J.F.M.; Verbakel, J.Y.; Christodoulou, E.; Vickers, A.J.; Roobol, M.J.; Steyerberg, E.W. Reporting and Interpreting Decision Curve Analysis: A Guide for Investigators. Eur. Urol. 2018, 74, 796–804. [Google Scholar] [CrossRef]
- Kerr, K.F.; Brown, M.D.; Zhu, K.; Janes, H. Assessing the Clinical Impact of Risk Prediction Models with Decision Curves: Guidance for Correct Interpretation and Appropriate Use. J. Clin. Oncol. 2016, 34, 2534–2540. [Google Scholar] [CrossRef]
- Irving, G.; Holden, J. The time-efficiency principle: Time as the key diagnostic strategy in primary care. Fam. Pract. 2013, 30, 386–389. [Google Scholar] [CrossRef]
- Manickam, K.; McClain, M.R.; Demmer, L.A.; Biswas, S.; Kearney, H.M.; Malinowski, J.; Massingham, L.J.; Miller, D.; Yu, T.W.; Hisama, F.M. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 2029–2037. [Google Scholar] [CrossRef]
- Buckley, J.R.; Kavarana, M.N.; Chowdhury, S.M.; Scheurer, M.A. Current Practice and Utility of Chromosome Microarray Analysis in Infants Undergoing Cardiac Surgery. Congenit. Heart Dis. 2015, 10, E131–E138. [Google Scholar] [CrossRef]
- Greenwood, R.D.; Rosenthal, A.; Parisi, L.; Fyler, D.C.; Nadas, A.S. Extracardiac abnormalities in infants with congenital heart disease. Pediatrics 1975, 55, 485–492. [Google Scholar]
- Copel, J.A.; Pilu, G.; Kleinman, C.S. Extracardiac anomalies and congenital heart disease. Semin. Perinatol. 1993, 17, 89–105. [Google Scholar] [PubMed]
- Tennstedt, C.; Chaoui, R.; Körner, H.; Dietel, M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: Results of a seven year necropsy study. Heart 1999, 82, 34–39. [Google Scholar] [CrossRef]
- Baker, K.; Sanchez-de-Toledo, J.; Munoz, R.; Orr, R.; Kiray, S.; Shiderly, D.; Clemens, M.; Wearden, P.; Morell, V.O.; Chrysostomou, C. Critical congenital heart disease--utility of routine screening for chromosomal and other extracardiac malformations. Congenit. Heart Dis. 2012, 7, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Dovjak, G.O.; Zalewski, T.; Seidl-Mlczoch, E.; Ulm, P.A.; Berger-Kulemann, V.; Weber, M.; Prayer, D.; Kasprian, G.J.; Ulm, B. Abnormal Extracardiac Development in Fetuses with Congenital Heart Disease. J. Am. Coll. Cardiol. 2021, 78, 2312–2322. [Google Scholar] [CrossRef]
- Hart, B.N.; Shubrook, J.H.; Mason, T. Pregestational Diabetes and Family Planning. Clin. Diabetes 2021, 39, 323–328. [Google Scholar] [CrossRef]
- Orós, M.; Perejón, D.; Serna, M.C.; Siscart, J.; Leon, J.; Ortega, M.; Salinas-Roca, B. Prevalence and risk factors of gestational diabetes in the health region of Lleida: A retrospective observational cohort study. J. Endocrinol. Investig. 2023, 46, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Gregory, E.C.; Ely, D.M. Trends and Characteristics in Gestational Diabetes: United States, 2016–2020. Natl. Vital Stat. Rep. 2022, 71, 1–15. [Google Scholar]
- Morris, C.A.; Braddock, S.R. Health Care Supervision for Children with Williams Syndrome. Pediatrics 2020, 145, e20193761. [Google Scholar] [CrossRef]
- Goldmuntz, E. 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 64–72. [Google Scholar] [CrossRef]
- Pepe, M.S.; Janes, H.; Longton, G.; Leisenring, W.; Newcomb, P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am. J. Epidemiol. 2004, 159, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Grunau, G.; Linn, S. Commentary: Sensitivity, Specificity, and Predictive Values: Foundations, Pliabilities, and Pitfalls in Research and Practice. Front. Public Health 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Alankarage, D.; Ip, E.; Szot, J.O.; Munro, J.; Blue, G.M.; Harrison, K.; Cuny, H.; Enriquez, A.; Troup, M.; Humphreys, D.T.; et al. Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet. Med. 2019, 21, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Mone, F.; Eberhardt, R.Y.; Morris, R.K.; Hurles, M.E.; McMullan, D.J.; Maher, E.R.; Lord, J.; Chitty, L.S.; Giordano, J.L.; Wapner, R.J.; et al. COngenital heart disease and the Diagnostic yield with Exome sequencing (CODE) study: Prospective cohort study and systematic review. Ultrasound Obstet. Gynecol. 2021, 57, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Saha, P.; Priest, J.R. Beyond Gene Panels: Whole Exome Sequencing for Diagnosis of Congenital Heart Disease. Circ. Genom. Precis. Med. 2018, 11, e002097. [Google Scholar] [CrossRef] [PubMed]
- Szot, J.O.; Cuny, H.; Blue, G.M.; Humphreys, D.T.; Ip, E.; Harrison, K.; Sholler, G.F.; Giannoulatou, E.; Leo, P.; Duncan, E.L.; et al. A Screening Approach to Identify Clinically Actionable Variants Causing Congenital Heart Disease in Exome Data. Circ. Genom. Precis. Med. 2018, 11, e001978. [Google Scholar] [CrossRef] [PubMed]
- Blue, G.M.; Humphreys, D.; Szot, J.; Major, J.; Chapman, G.; Bosman, A.; Kirk, E.P.; Sholler, G.F.; Harvey, R.P.; Dunwoodie, S.L.; et al. The promises and challenges of exome sequencing in familial, non-syndromic congenital heart disease. Int. J. Cardiol. 2017, 230, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Hays, T.; Hernan, R.; Disco, M.; Griffin, E.L.; Goldshtrom, N.; Vargas, D.; Krishnamurthy, G.; Bomback, M.; Rehman, A.U.; Wilson, A.T.; et al. Implementation of Rapid Genome Sequencing for Critically Ill Infants with Complex Congenital Heart Disease. Circ. Genom. Precis. Med. 2023, 16, 415–420. [Google Scholar] [CrossRef]
- Theis, J.L.; Olson, T.M. Whole Genome Sequencing in Hypoplastic Left Heart Syndrome. J. Cardiovasc. Dev. Dis. 2022, 9, 117. [Google Scholar] [CrossRef]
- Sweeney, N.M.; Nahas, S.A.; Chowdhury, S.; Batalov, S.; Clark, M.; Caylor, S.; Cakici, J.; Nigro, J.J.; Ding, Y.; Veeraraghavan, N.; et al. Rapid whole genome sequencing impacts care and resource utilization in infants with congenital heart disease. NPJ Genom. Med. 2021, 6, 29. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Cohort Descriptive Statistics |
|---|---|
| Sex | |
| Female | 443/1013 (43.7%) |
| Male | 570/1013 (56.3%) |
| Age at Consultation (Days) | Mean = 18.1 (SD = 47.0) Median = 3.0 (IQR: [1.0, 10.0]) |
| Age Group | |
| Neonate (0–28 days) | 880/1013 (86.9%) |
| Infant (29 days–1 year) | 133/1013 (13.1%) |
| Race/Ethnicity Group (Parent-Reported) | |
| Asian/Pacific Island | 25/1013 (2.5%) |
| Black/African American | 126/1013 (12.4%) |
| Hispanic/Latino | 110/1013 (10.9%) |
| Other | 3/1013 (0.3%) |
| White | 707/1013 (69.8%) |
| Unknown/Declined | 42/1013 (4.2%) |
| CHD Class | |
| APVR | 28/1013 (2.8%) |
| AVSD | 37/1013 (3.7%) |
| Complex | 132/1013 (13.0%) |
| Conotruncal | 249/1013 (24.6%) |
| Heterotaxy/Laterality Spectrum | 73/1013 (7.2%) |
| LVOTO | 260/1013 (25.7%) |
| RVOTO | 98/1013 (9.7%) |
| Septal | 136/1013 (13.4%) |
| ECA Status | |
| No | 580/1013 (57.3%) |
| Yes | 433/1013 (42.7%) |
| ECA Number | Mean = 1.0 (SD = 1.6) Median = 0 ([IQR: [0, 1.0]) Mode = 0 |
| Maternal Diabetes Status (Gestational & Pregestational) | |
| No/Unknown | 928/1013 (91.6%) |
| Yes | 85/1013 (8.4%) |
| Clinical Description at Evaluation | |
| Apparently Isolated/Non-Syndromic | 667/1013 (65.8%) |
| Possibly Syndromic CHD | 242/1013 (23.9%) |
| Confirmed Syndrome at Evaluation | 104/1013 (10.3%) |
| Consultation Time Period | |
| 2014–2018 | 313/1013 (50.9%) |
| 2019–2022 | 549/1013 (54.2%) |
| 2023 | 151/1013 (14.9%) |
| Genetic Testing Ordering Strategy | |
| None | 22/1013 (2.2%) |
| Prenatal Genetic Testing Only | 32/1013 (3.2%) |
| Outside Hospital Genetic Testing | 20/1013 (2.0%) |
| Targeted Cytogenetic Testing Only (FISH, karyotype) | 45/1013 (4.4%) |
| Chromosomal Microarray (postnatal) | 292/1013 (28.8%) |
| Targeted Molecular Genetic Testing Only (phenotype-specific/single-gene) | 12/1013 (1.2%) |
| Chromosome Microarray + Exome-Based Gene Panel/Exome Sequencing | 380/1013 (37.5%) |
| Genome Sequencing | 210/1013 (20.7%) |
| Number of Genetic Tests Completed | Mean = 1.5 Median = 1.0 (IQR: [1.0, 2.0]) Mode = 1.0 Range = 4.0 |
| Genetic Diagnosis Identified/Confirmed | |
| No | 769/1013 (75.9%) |
| Yes | 244/1013 (24.1%) |
| Genetic Testing Result Types | |
| Normal/Negative | 451/1013 (44.5%) |
| Inconclusive with ≥1 Variant(s) of Uncertain Significance | 319/1013 (31.5%) |
| Diagnostic | 243/1013 (24.0%) |
| Genetic Diagnosis Type(s) | |
| None/Unclear | 768/1013 (75.8%) |
| Cytogenetic | 168/1013 (16.6%) |
| Molecular Genetic | 70/1013 (6.9%) |
| Cytogenetic & Molecular (>1 Diagnosis) | 5/1013 (0.5%) |
| Clinical Diagnosis, with Uninformative Genetic Testing | 2/1013 (0.2%) ‡ |
| Extracardiac Anomalies Status | |||
|---|---|---|---|
| Variable | No | Yes | X2/Exact or Kruskal-Wallis Test p-Value |
| Sex | |||
| Female | 247/443 (55.8%) | 195/443 (44.2%) | p = 0.3951 |
| Male | 333/570 (58.4%) | 237/570 (41.6%) | |
| Age at Consultation (Days) | Mean = 13.4 (SD = 36.0) Median = 3.0 (IQR: [1.0, 8.0]) | Mean = 24.2 (SD = 58.0) Median = 2.0 (IQR: [1.0, 14.0]) | p = 0.5462 |
| Age Group | |||
| Neonate (0–28 days) | 520/880 (59.1%) | 360/880 (40.9%) | p = 0.0024 |
| Infant (29 days-1 year) | 60/133 (45.1%) | 73/133 (54.9%) | |
| Race/Ethnicity Group (Self-Reported) | |||
| Asian/Pacific Island | 13/25 (52.0%) | 12/25 (48.0%) | p = 0.2229 |
| Black/African American | 61/126 (48.4%) | 65/126 (51.6%) | |
| Hispanic/Latino | 62/110 (56.4%) | 48/110 (43.6%) | |
| Other | 1/3 (33.3%) | 2/3 (66.7%) | |
| White | 421/707 (59.6%) | 286/707 (40.5%) | |
| Unknown/Declined | 22/42 (52.4%) | 20/42 (47.6%) | |
| CHD Class | |||
| APVR | 23/28 (82.1%) | 5/28 (17.9%) | p < 0.0001 |
| AVSD | 17/37 (45.9%) | 20/37 (54.1%) | |
| Complex | 71/132 (53.8%) | 61/132 (46.2%) | |
| Conotruncal | 168/249 (67.5%) | 81/249 (32.5%) | |
| Heterotaxy/Laterality Spectrum | 8/73 (11.0%) | 65/73 (89.0%) | |
| LVOTO | 187/260 (71.9%) | 73/260 (28.1%) | |
| RVOTO | 71/98 (72.5%) | 27/98 (27.6%) | |
| Septal | 35/136 (25.7%) | 101/136 (74.3%) | |
| Maternal Diabetes Status (Gestational & Pregestational) | |||
| No/Unknown | 533/925 (57.6%) | 392/925 (42.4%) | p = 0.2850 |
| Yes | 44/85 (51.8%) | 41/85 (48.2%) | |
| Clinical Description at Evaluation | |||
| Apparently Isolated/Non-Syndromic | 526/667 (78.9%) | 141/667 (21.1%) | p < 0.0001 |
| Possibly Syndromic CHD | 27/242 (11.2%) | 215/242 (88.8%) | |
| Confirmed Syndrome at Evaluation | 27/104 (26.0%) | 77/104 (74.0%) | |
| Consultation Time Period | |||
| 2014–2018 | 195/313 (62.3%) | 118/313 (37.7%) | p = 0.0078 |
| 2019–2022 | 314/549 (57.2%) | 235/549 (42.8%) | |
| 2023 | 71/151 (47.0%) | 80/151 (53.0%) | |
| Genetic Testing Ordering Strategy | |||
| None | 19/22 (86.4%) | 3/22 (13.6%) | p < 0.0001 |
| Prenatal Genetic Testing Only | 6/32 (18.8%) | 26/32 (81.3%) | |
| Outside Hospital Genetic Testing | 2/20 (10.0%) | 18/20 (90.0%) | |
| Targeted Cytogenetic Testing Only (FISH, karyotype) | 17/45 (37.8%) | 28/45 (62.2%) | |
| Chromosomal Microarray (postnatal) | 203/292 (69.5%) | 89/292 (30.5%) | |
| Targeted Molecular Genetic Testing Only (phenotype-specific/single-gene) | 4/12 (33.3%) | 8/12 (66.7%) | |
| Chromosome Microarray + Exome-Based Gene Panel/Exome Sequencing | 217/380 (57.1%) | 163/380 (42.9%) | |
| Genome Sequencing | 112/210 (53.3%) | 98/210 (46.7%) | |
| Number of Genetic Tests Completed | Mean = 1.4 (SD = 0.6) Median = 1.0 (IQR: [1.0, 2.0]) Mode = 1.0 Range = 3.0 | Mean = 1.6 (SD = 0.7) Median = 2.0 (IQR: [1.0, 2.0]) Mode = 1.0 Range = 4.0 | p = 0.0001 |
| Genetic Diagnosis Identified/Confirmed | |||
| No | 502/769 (65.3%) | 267/769 (34.7%) | p < 0.0001 |
| Yes | 78/244 (32.0%) | 166/244 (68.0%) | |
| Genetic Testing Result Types | |||
| Normal/Negative | 296/451 (65.6%) | 155/451 (34.4%) | p < 0.0001 |
| Inconclusive with ≥1 Variant(s) of Uncertain Significance | 206/319 (64.6%) | 113/319 (35.4%) | |
| Diagnostic | 78/243 (32.1%) | 165/243 (67.9%) | |
| Genetic Diagnosis Type(s) | |||
| None/Unclear | 502/768 (65.4%) | 266/768 (34.6%) | Exact p < 0.0001 |
| Cytogenetic | 57/168 (33.9%) | 111/168 (66.1%) | |
| Molecular Genetic | 20/70 (28.6%) | 50/70 (71.4%) | |
| Cytogenetic & Molecular (>1 Diagnosis) | 1/5 (20.0%) | 4/5 (80.0%) | |
| Clinical Diagnosis, with Uninformative Genetic Testing | 0/2 (0.0%) | 2/2 (100.0%) | |
| Genetic Diagnosis Identified | |||
|---|---|---|---|
| Variable | No | Yes | X2/Exact or Kruskal-Wallis Test p-Value |
| Sex | |||
| Female | 321/443 (72.5% | 122/442 (27.5%) | p = 0.0235 |
| Male | 448/570 (78.6%) | 122/570 (21.4%) | |
| Age at Consultation (Days) | Mean = 17.0 (SD = 43.5) Median = 3.0 (IQR: [1.0, 10.0]) | Mean = 21.6 (SD = 56.6) Median = 2.0 (IQR: [1.0, 10.0]) | p = 0.0586 |
| Age Group | |||
| Neonate (0–28 days) | 671/880 (76.3%) | 209/880 (23.8%) | p = 0.5190 |
| Infant (29 days-1 year) | 98/133 (73.7%) | 35/133 (26.3%) | |
| Race/Ethnicity Group (Self-Reported) | |||
| Asian/Pacific Island | 19/25 (76.0%) | 6/25 (24.0%) | p = 0.8930 |
| Black/African American | 97/126 (77.0%) | 29/126 (23.0%) | |
| Hispanic/Latino | 84/110 (76.4%) | 26/110 (23.6%) | |
| Other | 2/3 (66.7%) | 1/3 (33.3%) | |
| White | 532/707 (75.3%) | 175/707 (24.8%) | |
| Unknown/Declined | 35/42 (83.3%) | 7/42 (16.7%) | |
| CHD Class | |||
| APVR | 24/28 (85.7%) | 4/28 (14.3%) | p < 0.0001 * |
| AVSD | 17/37 (45.9%) | 20/37 (54.1%) | |
| Complex | 99/132 (75.0%) | 33/132 (25.0%) | |
| Conotruncal | 186/249 (74.7%) | 63/249 (25.3%) | |
| Heterotaxy/Laterality Spectrum | 64/73 (87.7%) | 9/73 (12.3%) | |
| LVOTO | 212/260 (81.5%) | 48/260 (18.5%) | |
| RVOTO | 77/98 (78.6%) | 21/98 (21.4%) | |
| Septal | 90/136 (66.2%) | 46/136 (33.8%) | |
| ECA Status | |||
| No | 502/580 (86.6%) | 78/580 (13.5%) | p < 0.0001 |
| Yes | 267/433 (61.7%) | 166/433 (38.3%) | |
| Maternal Diabetes Status (Gestational & Pregestational) | |||
| No/Unknown | 694/925 (75.0%) | 231/925 (25.0%) | p = 0.0247 |
| Yes | 73/85 (85.9%) | 12/85 (14.1%) | |
| Clinical Description at Evaluation | |||
| Apparently Isolated/Non-Syndromic | 613/667 (91.9%) | 54/667 (8.1%) | p < 0.0001 |
| Possibly Syndromic CHD | 155/242 (64.1%) | 87/242 (36.0%) | |
| Confirmed Syndrome at Evaluation | 1/104 (0.96%) † | 103/104 (99.04%) | |
| Consultation Time Period | |||
| 2014–2018 | 256/313 (81.8%) | 57/313 (18.2%) | p = 0.0084 |
| 2019–2022 | 407/549 (74.1%) | 142/549 (25.9%) | |
| 2023 | 106/151 (70.2%) | 45/151 (29.8%) | |
| Genetic Testing Ordering Strategy | |||
| None | 21/22 (95.5%) | 1/22 (4.5%) | p < 0.0001 ** |
| Prenatal Genetic Testing Only | 11/32 (34.4%) | 21/32 (65.6%) | |
| Outside Hospital Genetic Testing | 5/20 (25.0%) | 15/20 (75.0%) | |
| Targeted Cytogenetic Testing Only (FISH, karyotype) | 7/45 (15.6%) | 38/45 (84.4%) | |
| Chromosomal Microarray (postnatal) | 231/292 (79.1%) | 61/292 (20.9%) | |
| Targeted Molecular Genetic Testing Only (phenotype-specific/single-gene) | 4/12 (33.3%) | 8/12 (66.7%) | |
| Chromosome Microarray + Exome-Based Gene Panel/Exome Sequencing | 323/380 (85.0%) | 57/380 (15.0%) | |
| Genome Sequencing | 167/210 (79.5%) | 43/210 (20.5%) | |
| Number of Genetic Tests Completed | Mean = 1.5 (SD = 0.7) Median = 1.0 (IQR: [1.0, 2.0]) Mode = 1.0 Range = 4.0 | Mean = 1.5 (SD = 0.7) Median = 1.0 (IQR: [1.0, 2.0]) Mode = 1.0 Range = 3.0 | p = 0.2799 |
| ECA Type | ECA Cohort Prevalence (%) | Genetic Diagnosis Identified (%) | Exact p-Value * | Odds Ratio (95% CI) ** |
|---|---|---|---|---|
| Skull | 1/1013 (0.1%) | 0/0 (0.0%) | p = 1.000 | |
| Head | 9/1013 (0.9%) | 4/9 (44.4%) | p = 0.2309 | |
| Neurological— Brain | 60/1013 (5.9%) | 33/60 (55.0%) | p < 0.0001 | 4.3 [2.5, 7.3] |
| Neurological— Other/Functional | 82/1013 (8.1%) | 40/82 (48.8%) | p < 0.0001 | 3.4 [2.1, 5.4] |
| Neural Tube Defect/Spinal Cord | 8/1013 (0.8%) | 4/8 (50.0%) | p = 0.1007 | |
| Eye | 29/1013 (2.9%) | 19/29 (66.5%) | p < 0.0001 | 6.4 [2.9, 14.0] |
| Ear | 4/1013 (0.4%) | 2/4 (50.0%) | p = 0.2462 | |
| Nose | 4/1013 (0.4%) | 2/4 (40.0%) | p = 1.000 | |
| Oral Cavity | 42/1013 (4.0%) | 23/42 (54.8%) | p < 0.0001 | 4.1 [2.2, 7.7] |
| Neck | 2/1013 (0.2%) | 2/2 (100.0%) | p = 0.0578 | |
| Throat—including Esophagus/Trachea | 34/1013 (3.4%) | 10/34 (29.4%) | p = 0.4228 | |
| Chest | 4/1013 (0.4%) | 1/4 (25.0%) | p = 1.000 | |
| Lung | 22/1013 (2.2%) | 10/22 (43.5%) | p = 0.0239 | 2.7 [1.2, 6.3] |
| Gastrointestinal & Abdominal Wall | 97/1013 (9.6%) | 34/97 (35.1%) | p = 0.0120 | 1.8 [1.2, 2.8] |
| Renal | 97/1013 (9.6%) | 33/97 (34.0%) | p = 0.0239 | 1.7 [1.1, 2.7] |
| Liver & Biliary | 45/1013 (4.4%) | 9/45 (20.0%) | p = 0.5956 | |
| Spleen | 45/1013 (4.4%) | 6/45 (13.3%) | p = 0.1032 | |
| Ribs & Vertebral | 38/1013 (3.8%) | 8/38 (21.0%) | p = 0.8467 | |
| Limb/Digit | 49/1013 (4.8%) | 24/49 (49.0%) | p = 0.0001 | 3.2 [1.8, 5.8] |
| Skin | 4/1013 (0.4%) | 3/4 (75.0%) | p = 0.0455 | 9.5 [1.0, 92.0] |
| Umbilical | 8/1013 (0.8%) | 1/8 (12.5%) | p = 0.6878 | |
| Immunologic | 3/1013 (0.3%) | 2/3 (66.7%) | p = 0.1458 | |
| Endocrine | 17/1013 (1.7%) | 10/17 (58.8%) | p = 0.0021 | 4.7 [1.8, 12.4] |
| Growth & Feeding | 75/1013 (7.4%) | 34/75 (45.3%) | p < 0.0001 | 2.9 [1.8, 4.6] |
| Hematologic | 9/1013 (0.9%) | 6/9 (66.7%) | p = 0.0080 | 6.4 [1.6, 25.9] |
| Other | 18/1013 (1.8%) | 6/18 (33.3%) | p = 0.4029 |
| Variable | Genetic Diagnosis (n) | Predictive Values (%) for ECA Status | Clinical Utility Index (CUI) for ECA Status (Positive and Negative) | |
|---|---|---|---|---|
| ECA Status | Yes | No | ||
| Yes | 166 | 267 | ECA(+) = 0.3834 (38.3%) | CUI(+) = 0.2608 * |
| No | 78 | 502 | ECA(−) = 0.8655 (86.6%) | CUI(−) = 0.5650 * |
| PSI = 0.2489 (24.9%) ** | SUI = 0.8258 † | |||
| Screening Metrics of ECA Status | ||||
| Sensitivity | 0.6803 | |||
| Specificity | 0.6528 | |||
| Accuracy | 0.6594 | |||
| Youden Index | 0.3331 | |||
| Number Needed to Diagnose (NND) | 3.00 | |||
| Evaluation of ECA Status as Predictor of Genetic Diagnoses | ||||
| Wald X2 (p-value) | Odds Ratio [95% CI] | AUC [95% CI] | Brier Score | |
| ECA Status (positive vs. negative) | 78.2 (p < 0.0001) | 4.0 [2.9, 5.4] | 0.667 [0.633, 0.700] | 0.168 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helm, B.M.; Ware, S.M. Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing. Genes 2024, 15, 505. https://doi.org/10.3390/genes15040505
Helm BM, Ware SM. Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing. Genes. 2024; 15(4):505. https://doi.org/10.3390/genes15040505
Chicago/Turabian StyleHelm, Benjamin M., and Stephanie M. Ware. 2024. "Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing" Genes 15, no. 4: 505. https://doi.org/10.3390/genes15040505
APA StyleHelm, B. M., & Ware, S. M. (2024). Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing. Genes, 15(4), 505. https://doi.org/10.3390/genes15040505