
Novel Pathogenic Variants Leading to Sporadic Amyotrophic Lateral Sclerosis in Greek Patients

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genetic Testing
2.3. In Silico Analysis
3. Results
3.1. SOD1
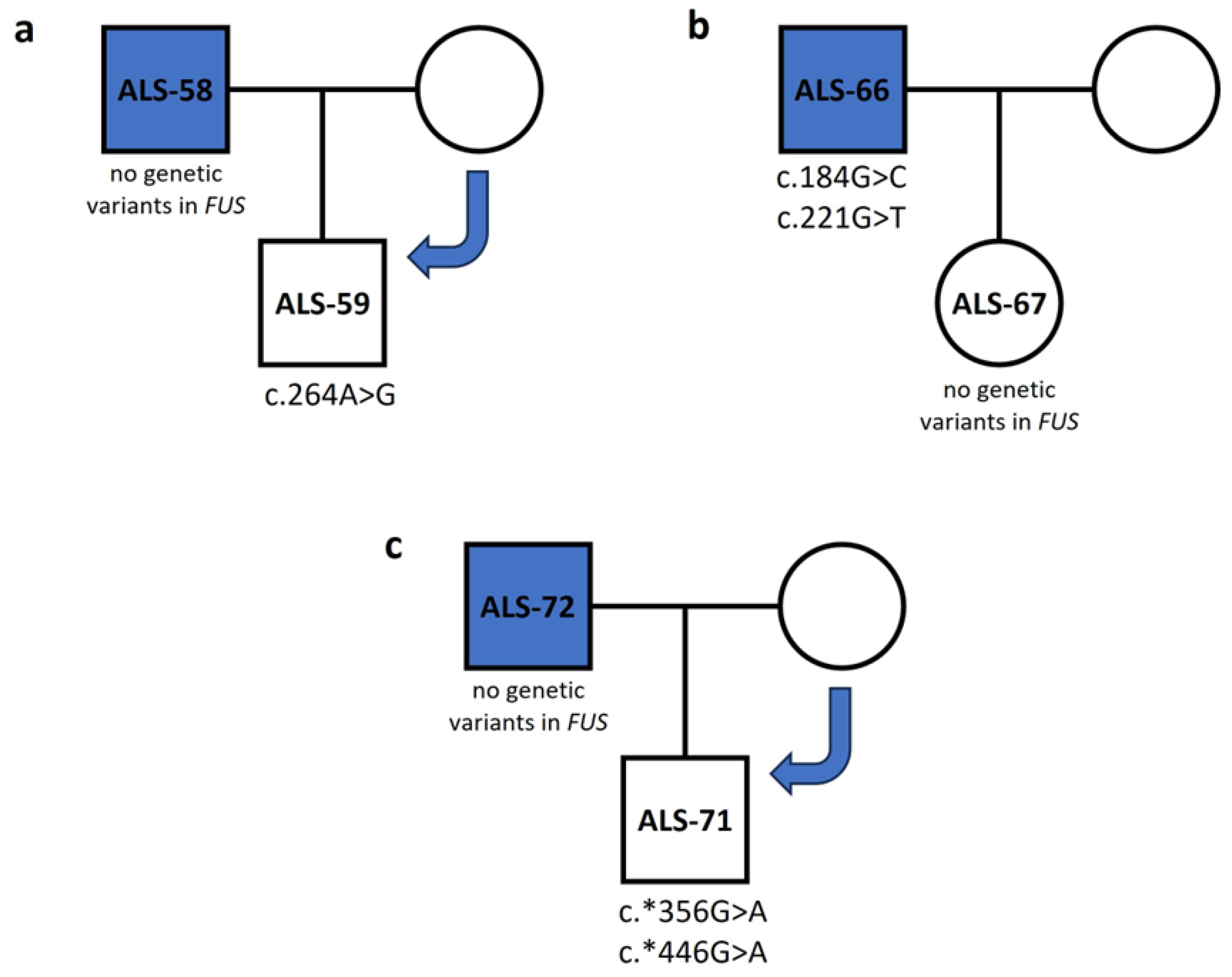
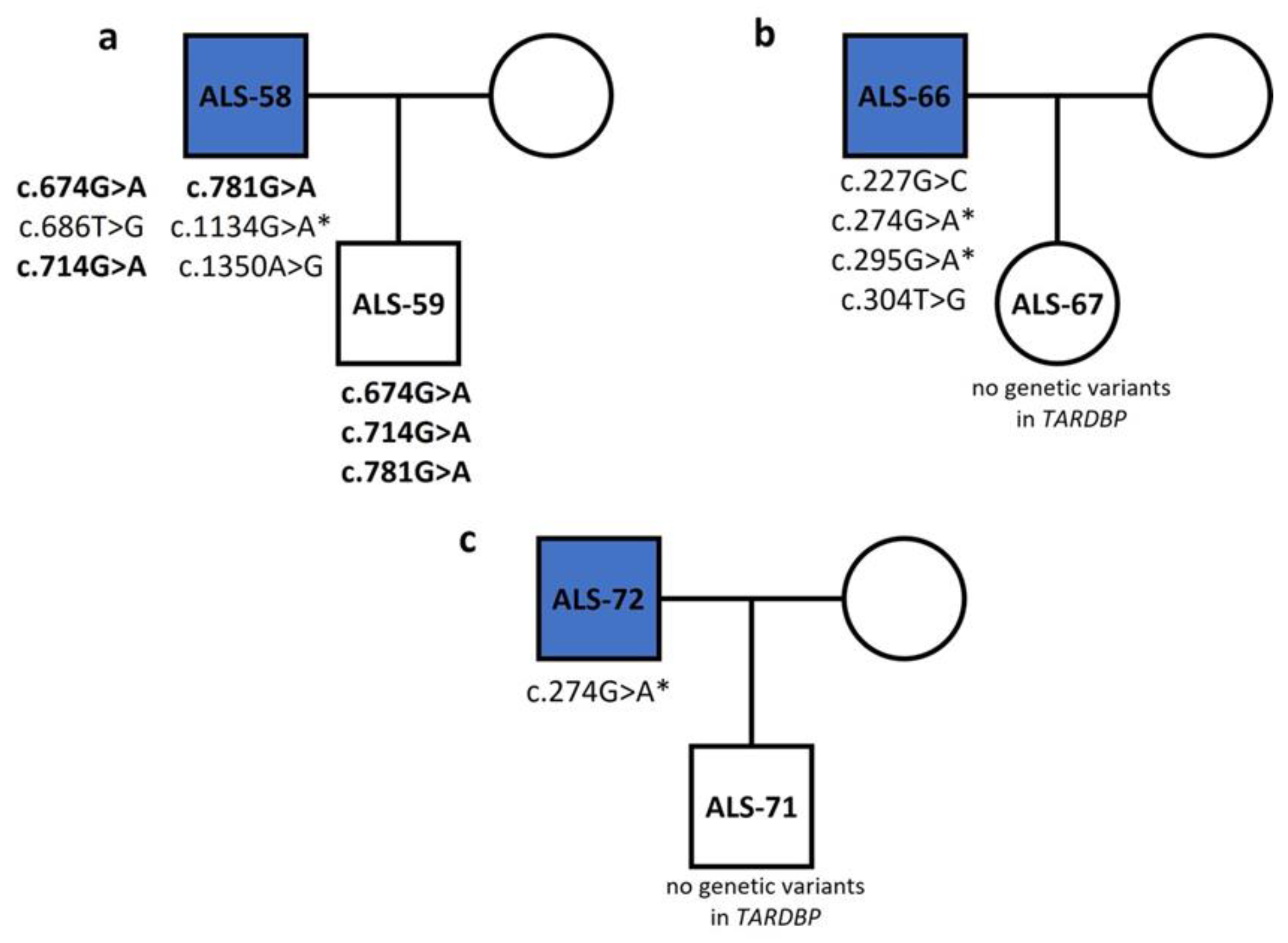
3.2. FUS
3.3. TARDBP
4. Discussion
Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gosset, P.; Camu, W.; Raoul, C.; Mezghrani, A. Prionoids in amyotrophic lateral sclerosis. Brain Commun. 2022, 4, fcac145. [Google Scholar] [CrossRef]
- Longinetti, E.; Regodón Wallin, A.; Samuelsson, K.; Press, R.; Zachau, A.; Ronnevi, L.O.; Kierkegaard, M.; Andersen, P.M.; Hillert, J.; Fang, F.; et al. The Swedish motor neuron disease quality registry. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Palese, F.; Sartori, A.; Verriello, L.; Ros, S.; Passadore, P.; Manganotti, P.; Barbone, F.; Pisa, F.E. Epidemiology of amyotrophic lateral sclerosis in Friuli-Venezia Giulia, North-Eastern Italy, 2002–2014: A retrospective population-based study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2019, 20, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Benjaminsen, E.; Alstadhaug, K.B.; Gulsvik, M.; Baloch, F.K.; Odeh, F. Amyotrophic lateral sclerosis in Nordland County, Norway, 2000–2015: Prevalence, incidence, and clinical features. Amyotroph. Lateral Scler. Frontotemporal Degener 2018, 19, 522–527. [Google Scholar] [CrossRef]
- Jun, K.Y.; Park, J.; Oh, K.W.; Kim, E.M.; Bae, J.S.; Kim, I.; Kim, S.H. Epidemiology of ALS in Korea using nationwide big data. J. Neurol. Neurosurg. Psychiatry 2019, 90, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Turgut, N.; Varol SaraÇoglu, G.; Kat, S.; Balci, K.; GÜldiken, B.; Birgili, O.; Kabayel, L. An epidemiologic investigation of amyotrophic lateral sclerosis in Thrace, Turkey, 2006–2010. Amyotroph. Lateral Scler. Frontotemporal Degener. 2019, 20, 100–106. [Google Scholar] [CrossRef]
- Zhou, S.; Zhou, Y.; Qian, S.; Chang, W.; Wang, L.; Fan, D. Amyotrophic lateral sclerosis in Beijing: Epidemiologic features and prognosis from 2010 to 2015. Brain Behav. 2018, 8, e01131. [Google Scholar] [CrossRef]
- Leighton, D.J.; Newton, J.; Stephenson, L.J.; Colville, S.; Davenport, R.; Gorrie, G.; Morrison, I.; Swingler, R.; Chandran, S.; Pal, S.; et al. Changing epidemiology of motor neurone disease in Scotland. J. Neurol. 2019, 266, 817–825. [Google Scholar] [CrossRef]
- Rose, L.; McKim, D.; Leasa, D.; Nonoyama, M.; Tandon, A.; Bai, Y.Q.; Amin, R.; Katz, S.; Goldstein, R.; Gershon, A. Trends in incidence, prevalence, and mortality of neuromuscular disease in Ontario, Canada: A population-based retrospective cohort study (2003–2014). PLoS ONE 2019, 14, e0210574. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Ji, A.L.; Zhang, X.; Chen, W.W.; Huang, W.J. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J. Med. Genet. 2017, 54, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Salem, K.; McCormick, M.L.; Wendlandt, E.; Zhan, F.; Goel, A. Copper-zinc superoxide dismutase-mediated redox regulation of bortezomib resistance in multiple myeloma. Redox Biol. 2015, 4, 23–33. [Google Scholar] [CrossRef]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Majzúnová, M.; Pakanová, Z.; Kvasnička, P.; Bališ, P.; Čačányiová, S.; Dovinová, I. Age-dependent redox status in the brain stem of NO-deficient hypertensive rats. J. Biomed. Sci. 2017, 24, 72. [Google Scholar] [CrossRef]
- Schmitt, N.D.; Agar, J.N. Parsing disease-relevant protein modifications from epiphenomena: Perspective on the structural basis of SOD1-mediated ALS. J. Mass. Spectrom. 2017, 52, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Xiao, X.; Li, M.; Ye, Z.; He, X.; Wei, J.; Zha, Y. FUS gene mutation in amyotrophic lateral sclerosis: A new case report and systematic review. Amyotroph. Lateral Scler. Frontotemporal Degener. 2024, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Deng, H.X.; Siddique, N.; Fecto, F.; Chen, W.; Yang, Y.; Liu, E.; Donkervoort, S.; Zheng, J.G.; Shi, Y.; et al. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010, 75, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Grassano, M.; Brodini, G.; De Marco, G.; Casale, F.; Fuda, G.; Salamone, P.; Brunetti, M.; Sbaiz, L.; Gallone, S.; Cugnasco, P.; et al. Phenotype analysis of fused in sarcoma mutations in amyotrophic lateral sclerosis. Neurol. Genet. 2022, 8, e200011. [Google Scholar] [CrossRef] [PubMed]
- Bäumer, D.; Hilton, D.; Paine, S.M.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.J.; Wang, D.; Wei, X.; Xia, X.G. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef]
- Assoni, A.F.; Foijer, F.; Zatz, M. Amyotrophic lateral sclerosis, FUS and Protein synthesis defects. Stem Cell Rev. Rep. 2023, 19, 625–638. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS–FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef]
- Mok, K.Y.; Koutsis, G.; Schottlaender, L.V.; Polke, J.; Panas, M.; Houlden, H. High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol. Aging 2012, 33, 1851.e1–1851.e15. [Google Scholar] [CrossRef]
- Mitropoulos, K.; Merkouri Papadima, E.; Xiromerisiou, G.; Balasopoulou, A.; Charalampidou, K.; Galani, V.; Zafeiri, K.V.; Dardiotis, E.; Ralli, S.; Deretzi, G.; et al. Genomic variants in the FTO gene are associated with sporadic amyotrophic lateral sclerosis in Greek patients. Hum. Genom. 2017, 11, 30. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Chen, Y.P.; Yu, S.H.; Wei, Q.Q.; Cao, B.; Gu, X.J.; Chen, X.P.; Song, W.; Zhao, B.; Wu, Y.; Sun, M.M. Role of genetics in amyotrophic lateral sclerosis: A large cohort study in Chinese mainland population. J. Med. Genet. 2020, 59, 840–849. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Steeves, M.; Bayrak-Toydemir, P.; Benson, K.A.; Coe, B.P.; Conlin, L.K.; Ganapathi, M.; Garcia, J.; Gollob, M.H.; Jobanputra, V.; et al. Recommendations for risk allele evidence curation, classification, and reporting from the ClinGen Low Penetrance/Risk Allele Working Group. Genet. Med. 2023, 26, 101036. [Google Scholar] [CrossRef]
- Battistini, S.; Giannini, F.; Greco, G.; Bibbò, G.; Ferrera, L.; Marini, V.; Causarano, R.; Casula, M.; Lando, G.; Patrosso, M.C.; et al. SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J. Neurol. 2005, 252, 782–788. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Eisen, A.; Mezei, M.M.; Stewart, H.G.; Fabros, M.; Gibson, G.; Andersen, P.M. SOD1 gene mutations in ALS patients from British Columbia, Canada: Clinical features, neurophysiology and ethical issues in management. Amyotroph. Lateral Scler. 2008, 9, 108–119. [Google Scholar] [CrossRef]
- Alavi, A.; Nafissi, S.; Rohani, M.; Zamani, B.; Sedighi, B.; Shamshiri, H.; Fan, J.B.; Ronaghi, M.; Elahi, E. Genetic analysis and SOD1 mutation screening in Iranian amyotrophic lateral sclerosis patients. Neurobiol. Aging 2013, 34, 1516.e1–1516.e8. [Google Scholar] [CrossRef]
- Andersen, P.M.; Sims, K.B.; Xin, W.W.; Kiely, R.; O’Neill, G.; Ravits, J.; Pioro, E.; Harati, Y.; Brower, R.D.; Levine, J.S.; et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2003, 4, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, S.; Udagawa, T.; Fujioka, Y.; Honda, D.; Okado, H.; Watanabe, H.; Katsuno, M.; Ishigaki, S.; Sobue, G. 3′UTR length-dependent control of synGAP isoform α2 mRNA by FUS and ELAV-like proteins promotes dendritic spine maturation and cognitive function. Cell Rep. 2017, 20, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.Y.; Peng, Y.; Feng, X.H.; Wang, X.N.; Sun, Q.; Liu, M.S.; Li, X.G.; Cui, L.Y. Screening of the FUS gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. Eur. J. Neurol. 2012, 19, 977–983. [Google Scholar] [CrossRef]
- Ticozzi, N.; Silani, V.; LeClerc, A.L.; Keagle, P.; Gellera, C.; Ratti, A.; Taroni, F.; Kwiatkowski, T.J., Jr.; McKenna-Yasek, D.M.; Sapp, P.C.; et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 2009, 73, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; van Es, M.A.; van Vught, P.W.; Spliet, W.G.; van Engelen-Lee, J.; de Visser, M.; Wokke, J.H.; Schelhaas, H.J.; Ophoff, R.A.; Fumoto, K.; et al. FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch. Neurol. 2010, 67, 224–230. [Google Scholar] [CrossRef]
- Dini Modigliani, S.; Morlando, M.; Errichelli, L.; Sabatelli, M.; Bozzoni, I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA–FUS regulatory circuitry. Nat. Commun. 2014, 5, 4335. [Google Scholar] [CrossRef]
- Drepper, C.; Herrmann, T.; Wessig, C.; Beck, M.; Sendtner, M. C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol. Aging 2011, 32, 548.e1–548.e4. [Google Scholar] [CrossRef]
- Sabatelli, M.; Moncada, A.; Conte, A.; Lattante, S.; Marangi, G.; Luigetti, M.; Lucchini, M.; Mirabella, M.; Romano, A.; Del Grande, A.; et al. Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4748–4755. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Labbé, C.; Rayaprolu, S.; Soto-Ortolaza, A.; Ogaki, K.; Uitti, R.J.; Wszolek, Z.K.; Ross, O.A. Investigating FUS variation in Parkinson’s disease. Park. Relat. Disord. 2014, 20 (Suppl. 1), S147–S149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Sephton, C.F.; Tang, A.A.; Kulkarni, A.; West, J.; Brooks, M.; Stubblefield, J.J.; Liu, Y.; Zhang, M.Q.; Green, C.B.; Huber, K.M.; et al. Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, E4769–E4778. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.; Corrado, L.; Piola, B.; Comi, C.; Cantello, R.; D’Alfonso, S.; Mazzini, L.; De Marchi, F. Variability in Clinical Phenotype in TARDBP Mutations: Amyotrophic Lateral Sclerosis Case Description and Literature Review. Genes 2023, 14, 2039. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef]
- Lattante, S.; Sabatelli, M.; Bisogni, G.; Marangi, G.; Doronzio, P.N.; Martello, F.; Renzi, A.G.; Del Giudice, E.; Leon, A.; Cimbolli, P.; et al. Evaluating the contribution of the gene TARDBP in Italian patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2023, 30, 1246–1255. [Google Scholar] [CrossRef]
- Saxton, A.D.; Kraemer, B.C. Human Ubiquilin 2 and TDP-43 copathology drives neurodegeneration in transgenic Caenorhabditis elegans. G3 2021, 11, jkab158. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| cDNA | Exon | rsID | Amino Acid Change | Functional Protein | Type of Variant | MutationTaster | PolyPhen-2 | SIFT | Samples (Genotype) |
|---|---|---|---|---|---|---|---|---|---|
| c.248C>G | 3 | - | - | Not included | Silent | Disease causing | - | - | ALS-12 (het) |
| c.251T>G | 3 | - | p.C57W | Not included | Missense | Disease causing | Probably damaging | Affects protein function | ALS-12 (het) |
| c.254C>A | 3 | rs549580868 | - | Not included | Silent | Disease causing | - | - | ALS-12 (het) |
| c.292A>T | 3 | - | p.H71L | Not included | Missense | Disease causing | Probably damaging | Affects protein function | ALS-22 (het) |
| c.308T>C | 3 | - | - | Not included | Silent | Disease causing | - | - | ALS-22 (het) |
| c.349A>C | 4 | - | p.D90A | p.D11A | Missense | Disease causing | Benign | Tolerated | ALS-39 (hom) |
| cDNA | Exon | Protein | Type of Variant | rsID | MutationTaster | PolyPhen-2 | SIFT | Samples (Genotype) |
|---|---|---|---|---|---|---|---|---|
| c.101C>A | 2 | p.Q8K | Missense | - | Disease causing | Benign | Affects protein function | ALS-58 (het) |
| c.162G>T | 3 | p.S28I | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-30 (het) |
| c.184G>C | 3 | p.Q35H | Missense | rs772271532 | Disease causing | Probably damaging | Affects protein function | ALS-66 (het) |
| c.221G>T | 3 | p.G48C | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-66 (het) |
| c.223C>A | 3 | - | Silent | - | Polymorphism | - | - | ALS-9 (het), ALS-12 (hom), ALS-13 (hom), ALS-14 (hom), ALS-21 (hom), ALS-24 (het), ALS-30 (het), ALS-38 (hom), ALS-40 (het), ALS-68 (het) |
| c.626A>C | 6 | p.M183L | Missense | rs762914131 | Disease causing | Benign | Affects protein function | ALS-30 (het), ALS-38 (het), ALS-57 (het) |
| c.759G>T | 6 | p.G227V | Missense | - | Disease causing | Benign | Affects protein function | ALS-70 (het) |
| c.760C>G | 6 | - | Silent | rs151073460 | Disease causing | - | - | ALS-10 (het), ALS-30 (het) |
| c.800A>T | 6 | p.R241* | Nonsense | - | Disease causing | - | - | ALS-38 (het), ALS-40 (het) |
| c.806C>T | 6 | p.R243C | Missense | rs1165095258 | Disease causing | Benign | Affects protein function | ALS-2 (het), ALS-10 (het), ALS-22 (het), ALS-25 (het), ALS-30 (het) |
| c.830G>T | 6 | p.G251C | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-65 (het) |
| c.*41G>A | 3′ UTR | - | - | rs80301724 | Disease causing | - | - | ALS-13 (het) |
| c.*81C>T | 3′ UTR | - | - | rs768544815 | Polymorphism | - | - | ALS-73 (het) |
| c.*306T>C | 3′ UTR | - | - | - | Polymorphism | - | - | ALS-63 (het) |
| c.*354A>T | 3′ UTR | - | - | - | Polymorphism | - | - | ALS-70 (het) |
| c.*356G>A | 3′ UTR | - | - | rs886051940 | Polymorphism | - | - | ALS-60 (het), ALS-61 (het), ALS-64 (het), ALS-65 (het), ALS-69 (het) |
| c.*362T>G | 3′ UTR | - | - | - | Polymorphism | - | - | ALS-73 (het) |
| c.*370A>T | 3′ UTR | - | - | - | - | - | - | ALS-69 (het) |
| c.*406G>A | 3′ UTR | - | - | - | - | - | - | ALS-69 (het) |
| cDNA | Exon | Protein | Type of Variant | rsID | MutationTaster | PolyPhen-2 | SIFT | Samples (Genotype) |
|---|---|---|---|---|---|---|---|---|
| c.5T>C | 1 (5′ UTR) | - | - | - | Disease causing | - | - | ALS-6 (het) |
| c.24C>G | 1 (5′ UTR) | - | - | rs965172966 | Polymorphism | - | - | ALS-2 (het), ALS-6 (het), ALS-13 (hom) |
| c.227G>C | 2 | p.R41P | Missense | - | Disease causing | Benign | Affects protein function | ALS-66 (het) |
| c.274G>A | 2 | p.E57K | Missense | - | Disease causing | Benign | Affects protein function | ALS-30 (het), ALS-40 (het), ALS-41 (het), ALS-66 (het), ALS-68 (het), ALS-69 (het), ALS-72 (het), ALS-73 (het) |
| c.295G>A | 2 | p.D64N | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-41 (het), ALS-66 (het) |
| c.295G>T | 2 | p.D64Y | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-57 (het) |
| c.300T>C | 2 | - | Silent | rs61730366 | Disease causing | - | - | ALS-63 (het) |
| c.303C>G | 2 | - | Silent | - | Disease causing | - | - | ALS-57 (het) |
| c.304T>G | 2 | p.W67G | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-66 (het) |
| c.363G>A | 3 | - | Silent | - | Disease causing | - | - | ALS-73 (het) |
| c.405G>A | 3 | - | Silent | - | Disease causing | - | - | ALS-73 (het) |
| c.468G>T | 3 | p.E121D | Missense | - | Polymorphism | Benign | Tolerated | ALS-73 (het) |
| c.487G>A | 3 | p.E128K | Missense | - | Disease causing | Possibly damaging | Tolerated | ALS-73 (het) |
| c.490G>T | 3 | p.V129F | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-73 (het) |
| c.500T>G | 3 | p.V132G | Missense | rs766116483 | Disease causing | Possibly damaging | Affects protein function | ALS-25 (het) |
| c.594G>C | 4 | p.Q163H | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-41 (het) |
| c.674G>A | 5 | p.R190K | Missense | - | Disease causing | Benign | Tolerated | ALS-14 (het), ALS-58 (het) |
| c.686T>G | 5 | p.F193L | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-58 (het) |
| c.703G>A | 5 | p.D200N | Missense | - | Disease causing | Possibly damaging | Tolerated | ALS-57 (het) |
| c.714G>A | 5 | - | Silent | rs1333943256 | Disease causing | - | - | ALS-58 (het) |
| c.715G>A | 5 | p.D204N | Missense | - | Disease causing | Benign | Tolerated | ALS-57 (het) |
| c.741G>T | 5 | p.Q212H | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-14 (het) |
| c.744C>G | 5 | p.Y214* | Nonsense | - | Disease causing | - | - | ALS-57 (het) |
| c.777A>T | 5 | - | Silent | - | Disease causing | - | - | ALS-57 (het) |
| c.781A>G | 5 | p.R226G | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-57 (het), ALS-58 (het) |
| c.801A>T | 5 | - | Silent | - | Disease causing | - | - | ALS-57 (het) |
| c.972T>A | 6 | - | Silent | - | Disease causing | - | - | ALS-70 (het) |
| c.995G>T | 6 | p.G297V | Missense | rs1643653768 | Disease causing | Benign | Affects protein function | ALS-63 (het) |
| c.1001G>A | 6 | p.G299E | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-70 (het) |
| c.1134G>A | 6 | - | Silent | - | Disease causing | - | - | ALS-9 (het), ALS-13 (het), ALS-23 (het), ALS-25 (het), ALS-38 (het), ALS-58 (het), ALS-68 (het), ALS-73 (het) |
| c.1180C>A | 6 | p.Q326R | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-70 (het) |
| c.1182G>A | 6 | - | Silent | - | Disease causing | - | - | ALS-70 (het) |
| c.1322C>T | 6 | p.S406F | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-21 (het) |
| c.1326G>T | 6 | p.K407N | Missense | - | Disease causing | Probably damaging | Affects protein function | ALS-70 (het) |
| c.1328C>T | 6 | p.S408F | Missense | - | Disease causing | Possibly damaging | Affects protein function | ALS-21 (het) |
| c.1347G>A | 6 (3′ UTR) | - | - | Disease causing | - | - | ALS-68 (het) | |
| c.1350A>G | 6 (3′ UTR) | - | - | Polymorphism | - | - | ALS-58 (het) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivantsik, O.; John, A.; Kydonopoulou, K.; Mitropoulos, K.; Gerou, S.; Ali, B.R.; Patrinos, G.P. Novel Pathogenic Variants Leading to Sporadic Amyotrophic Lateral Sclerosis in Greek Patients. Genes 2024, 15, 309. https://doi.org/10.3390/genes15030309
Ivantsik O, John A, Kydonopoulou K, Mitropoulos K, Gerou S, Ali BR, Patrinos GP. Novel Pathogenic Variants Leading to Sporadic Amyotrophic Lateral Sclerosis in Greek Patients. Genes. 2024; 15(3):309. https://doi.org/10.3390/genes15030309
Chicago/Turabian StyleIvantsik, Ouliana, Anne John, Kyriaki Kydonopoulou, Konstantinos Mitropoulos, Spyridon Gerou, Bassam R. Ali, and George P. Patrinos. 2024. "Novel Pathogenic Variants Leading to Sporadic Amyotrophic Lateral Sclerosis in Greek Patients" Genes 15, no. 3: 309. https://doi.org/10.3390/genes15030309
APA StyleIvantsik, O., John, A., Kydonopoulou, K., Mitropoulos, K., Gerou, S., Ali, B. R., & Patrinos, G. P. (2024). Novel Pathogenic Variants Leading to Sporadic Amyotrophic Lateral Sclerosis in Greek Patients. Genes, 15(3), 309. https://doi.org/10.3390/genes15030309